一種大黃蟄蟲丸的指紋圖譜檢測方法與流程
2023-05-27 02:50:46 2
本發明涉及一種中藥製劑的指紋圖譜檢測方法,具體涉及大黃蟄蟲丸的指紋圖譜檢測方法。
背景技術:
大黃蟄蟲丸由熟大黃、土鱉蟲(炒)、水蛭(制)、虻蟲(去翅足,炒)、蠐螬(炒)、乾漆(煅)、桃仁、苦杏仁(炒)、黃芩、地黃、白芍、甘草十二味中藥製成,具有活血破瘀,通經消痞的藥理作用。該複方藥味複雜,含有黃酮類、蒽衍生物類、苯丁酮、苷類、鞣質類等主要化學成分。大黃蟄蟲丸含有的藥物成分比較多,既有極性小的蒽醌類成分,又有糖苷、鞣質等極性大的化合物,保留時間相差較大,現有技術的分析方法不能全面、客觀的檢測和評價大黃蟄蟲丸的質量。
技術實現要素:
發明目的:本發明的目的是解決現有技術的不足,提供一種大黃蟄蟲丸的指紋圖譜檢測方法,該檢測方法可以客觀、全面、準確的評價大黃蟄蟲丸的質量,對控制大黃蟄蟲丸的內在質量與保證臨床療效具有重要意義。
技術方案:為了實現以上技術目的,本發明採取的技術方案為:
一種大黃蟄蟲丸的指紋圖譜檢測方法,該指紋圖譜檢測方法包括以下步驟:
(1)供試品溶液製備:取大黃蟄蟲丸粉末,精密稱定,置圓底燒瓶中,精密加入甲醇或乙醇,稱定重量,超聲提取,放冷,再稱定重量,用甲醇補足減失的重量,搖勻,離心取上清液,用0.22μm微孔濾膜過濾取濾液,製成供試品溶液;
(2)對照品溶液的製備:精密稱取芍藥苷、蘆薈大黃素、大黃酸、大黃素、大黃酚、大黃素甲醚、毛蕊花糖苷對照品,用甲醇或乙醇溶解,分別配置成適宜濃度的對照品儲備液;
(3)高效液相色譜法測定本品的對照指紋圖譜:取5~10批大黃蟄蟲丸供試品在適當的色譜條件下進樣,測得各批次供試品的指紋圖譜,以這些指紋圖譜為基礎,用相似度軟體計算獲得共有模式作為本品的對照指紋圖譜;
(4)取對照品溶液按步驟(3)相同的色譜條件進樣,測得各對照品溶液的高效液相圖譜;
(5)高效液相色譜法測定供試品溶液指紋圖譜:取步驟(1)供試品溶液按步驟(3)相同的色譜條件進樣,測得供試品溶液的指紋圖譜;並根據步驟(4)所得對照品溶液的高效液相圖譜歸屬各色譜峰;
(6)將步驟(5)得到的供試品溶液指紋圖譜和步驟(3)得到的對照指紋圖譜用相似度軟體評價。
作為優選方案,以上所述的大黃蟄蟲丸的指紋圖譜檢測方法,步驟(3)色譜柱:Thermo C18色譜柱(規格)流動相:A相甲醇-B相0.1%甲酸水溶液,梯度洗脫;流速1.0ml/min;檢測波長270nm;柱溫:30℃;進樣量10μL。
作為更優選方案,以上所述的大黃蟄蟲丸的指紋圖譜檢測方法,步驟(3)梯度洗脫程序如下表:
作為更優選方案,以上所述的大黃蟄蟲丸的指紋圖譜檢測方法,步驟(1)中供試品溶液製備方法為:取大黃蟄蟲丸粉末1g,精密稱定,置圓底燒瓶中,精密加入甲醇25mL,稱定重量,超聲提取40min,再稱定重量,用甲醇補足減失的重量,搖勻,離心取上清液,用0.22μm微孔濾膜過濾取濾液,製成供試品溶液。
作為更優選方案,以上所述的大黃蟄蟲丸的指紋圖譜檢測方法,其特徵在於,步驟(2)對照品溶液的製備:精密稱取芍藥苷、蘆薈大黃素、大黃酸、大黃素、大黃酚、大黃素甲醚和毛蕊花糖苷對照品各2g,用甲醇溶解,分別配置成1mg/mL的對照品儲備液。
作為優選方案,以上所述的大黃蟄蟲丸的指紋圖譜檢測方法,大黃蟄蟲丸由熟大黃300g、土鱉蟲(炒)30g、水蛭(制)60g、虻蟲(去翅足,炒)45g、蠐螬(炒)45g、乾漆(煅)30g、桃仁120g、苦杏仁(炒)120g、黃芩60g、地黃300g、白芍120g和甘草90g製成。如選用北京同仁堂股份有限公司同仁堂製藥廠生產的國藥準字Z11020002的大黃蟄蟲丸。以上指紋圖譜Ⅰ的色譜條件優化實驗:
色譜條件的篩選:
一、色譜柱選擇
本發明分別考察了Thermo C18色譜柱((250mm×4.6mm i.d.,5μm)、Tigerkin C18柱(250mm×4.6mm i.d.,5μm)、Waters Atiantis T3(250mm×4.6mm i.d.,5μm)色譜柱對大黃蟄蟲丸的分離效果。結果表明,Thermo C18色譜柱((250mm×4.6mm i.d.,5μm)柱對極性差異較大的化合物有很好的保留,故選擇Thermo C18色譜柱((250mm×4.6mm i.d.,5μm)為指紋圖譜分析用色譜柱。
二、檢測波長選擇
本發明對大黃蟄蟲丸的甲醇提取液進行液相全波長掃描,在200nm~400nm處均有顯著的吸收,在270nm處吸收最為強烈,因此採用HPLC/UV方法270nm進行分析。
三、流動相體系選擇
由於大黃蟄蟲丸含有的由於活性成分比較多,既有極性小的黃酮類、蒽醌類成分,又有糖苷、鞣質等極性大的化合物,保留時間相差較大,為了保證在合適的分析時間內色譜峰達到較好的分離,本發明分別考察了甲醇-水、甲醇-0.05﹪甲酸溶液、甲醇-0.1﹪甲酸、甲醇-0.05﹪磷酸、甲醇-0.1﹪磷酸溶液對分離的影響,結果表明甲酸濃度0.1﹪時色譜峰峰型對稱、分離良好,故選擇甲醇-0.1﹪甲酸溶液(V/V)為色譜分離用流動相體系。
四、柱溫選擇
本發明分別考察了25℃、30℃、35℃、40℃和45℃柱溫條件下對分離的影響,結果:柱溫在30℃時,各成分分離度較好。色譜分析柱溫選擇30℃。
五、流速的選擇
本發明分別考察了0.5ml/min、0.7ml/min、0.8ml/min、0.9ml/min、1.0ml/min、1.2ml/min的流速對分離的影響,結果:以1.0ml/min的流速分離效果較佳。
六、流動相梯度選擇
本發明考察了不同梯度洗脫條件對分離的影響,結果以下述優選的流動相梯度為佳,故選定該梯度為指紋圖譜分析的色譜條件,
以上指紋圖譜Ⅰ的方法學驗證
1.1精密度實驗
取同一供試品溶液在上述優選色譜條件下連續測定6次,計算指紋峰的保留時間和峰面積的RSD值,相對保留時間RSD分別為0.18%、0.19%、0.16%、0.12%、0.10%、0.13%、0.12%、0.12%、0.11%,各色譜峰相對保留時間穩定,各主要色譜峰相對峰面積基本一致,RSD小於3%
1.2重複性實驗
平行取6份供試品溶液,按上述優選色譜條件進樣測定,計算指紋峰的保留時間和峰面積的RSD值,結果相對保留時間RSD為0.39%、0.37%、0.37%、0.31%、0.27%、0.25%,峰面積RSD值1.67%、1.57%、2.50%、1.30%、1.57%、1.67%。
1.3穩定性實驗
取同一供試品溶液在上述條件下,分別在0、2、4、8、12、24h測定其指紋圖譜,各色譜峰保留時間RSD分別是0.38%、0.46%、0.49%、0.44%、0.43%、0.46%、0.39%、0.37%、0.35%各主要色譜峰,相對峰面積RSD分別是2.85%、2.60%、2.68%、2.53%、2.43%、2.99%、2.24%、2.25%、2.42%。
1.4延長衝洗實驗
取供試品溶液進樣,在梯度結束後以甲醇衝洗1h,沒有出現其他色譜峰。
本發明將供試品指紋圖譜與對照指紋圖譜經計算機相似度評價軟體(選用中國藥典委推薦的「中藥指紋圖譜計算機輔助相似度評價軟體2004A版」)計算,獲得指紋圖譜相似度數值,依據相似度數值,應用於大黃蟄蟲丸的質量控制,以指紋圖譜相似度應大於0.90為檢驗質控標準。本發明提供的大黃蟄蟲丸的指紋圖譜檢測方法用於生產檢驗質量控制,所得產品具有較高的質量穩定性,可確保產品安全有效。
有益效果:本發明提供的中藥大黃蟄蟲丸的指紋圖譜檢測方法具有以下優點:
本發明針對現有技術檢測方法的諸多不足,通過大量實驗篩選出最佳的流動相組成,檢測波長、色譜柱,柱溫等分析條件,經多次實驗驗證表明,本發明提供的檢測方法,可建立良好的指紋圖譜。採用本發明提供的大黃蟄蟲丸指紋圖譜檢測方法,可以同時檢測大黃蟄蟲丸中極性差異較大的多種活性成分,從而可以全面、客觀、準確的檢測和評價大黃蟄蟲丸的質量,為保證臨床療效具有重要意義。
附圖說明
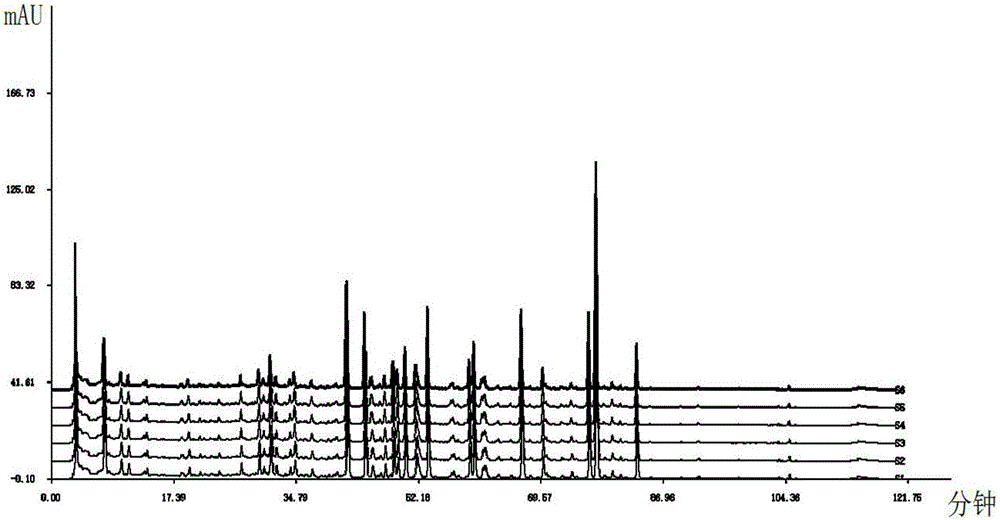
圖1為本發明大黃蟄蟲丸的對照指紋圖譜。
圖2為6個批次大黃蟄蟲丸的指紋圖譜。
具體實施方式
下面結合具體實施例進一步闡明本發明,應理解這些實施例僅用於說明本發明而不用於限制本發明的範圍,在閱讀了本發明之後,本領域技術人員對本發明的各種等價形式的修改均落於本申請所附權利要求所限定的範圍。
實施例1
一種大黃蟄蟲丸的指紋圖譜檢測方法,所述指紋圖譜檢測方法,包括以下步驟:
(1)供試品溶液製備:取大黃蟄蟲丸粉末1g,精密稱定,置圓底燒瓶中,精密加入甲醇25mL,稱定重量,超聲提取45min,放冷,再稱定重量,用甲醇補足減失的重量,搖勻,離心取上清液,用0.22μm微孔濾膜過濾取濾液,製成供試品溶液;
(2)對照品溶液的製備:精密稱取芍藥苷、蘆薈大黃素、大黃酸、大黃素、大黃酚、大黃素甲醚、毛蕊花糖苷對照品各2g,用甲醇溶解,分別配置成1mg/mL的對照品儲備液;
(3)高效液相色譜法測定本品的對照指紋圖譜:取6批大黃蟄蟲丸供試品在適當的色譜條件下進樣,測得各批次供試品的指紋圖譜,以這些指紋圖譜為基礎,用相似度軟體計算獲得共有模式作為本品的對照指紋圖譜(如圖1所示);
(4)取對照品溶液按步驟(3)相同的色譜條件進樣,測得各對照品溶液的高效液相圖譜;
(5)高效液相色譜法測定供試品溶液指紋圖譜:取步驟(1)供試品溶液按步驟(3)相同的色譜條件進樣,測得供試品溶液的指紋圖譜(如如圖2所示);並根據步驟(4)所得對照品溶液的高效液相圖譜歸屬各色譜峰(圖2中峰1為黃芩苷,峰2為黃芩素,峰3為大黃酸,峰4為大黃素,峰5為大黃酚,峰6為大黃素甲醚);
(6)將步驟(5)得到的供試品溶液指紋圖譜和步驟(3)得到的對照指紋圖譜用相似度軟體評價(如表1)。
以上所述的大黃蟄蟲丸的指紋圖譜檢測方法,步驟(3)色譜柱:Thermo C18色譜柱(規格)流動相:A相甲醇-B相0.1%甲酸水溶液,梯度洗脫;流速1.0ml/min;檢測波長270nm;柱溫:30℃;進樣量10μL。梯度洗脫程序如下表:
表1 6批大黃蟄蟲丸指紋圖譜相似度評價結果
通過以上實驗結果表明,本發明提供的大黃蟄蟲丸的指紋圖譜檢測方法,可以客觀,全面,有針對性的分析和評價大黃蟄蟲丸成品的質量,對保證臨床療效具有重要意義,本發明提供的指紋圖譜檢測方法,較現有技術單類成分分析,取得了明顯的技術進步。
以上所述僅是本發明的優選實施方式,應當指出,對於本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護範圍。