一種抗腫瘤藥物的新晶型及其製備方法和用途與流程
2023-12-10 07:26:37 2
本發明涉及化學製藥領域,尤其涉及一種抗腫瘤藥物palbociclib的晶型及其製備方法和用途。
背景技術:
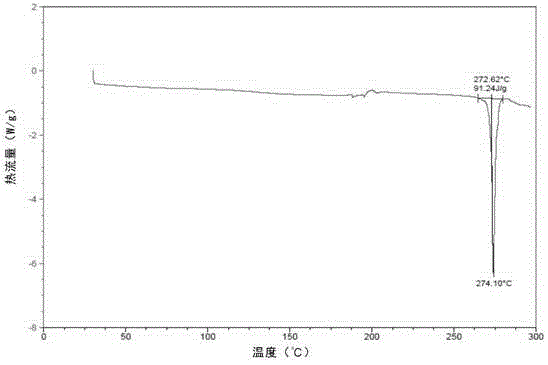
:輝瑞公司研發,目前用於治療乳腺癌的palbociclib的化學名為6-乙醯基-8-環戊基-5-甲基-2-{[5-(哌嗪-1-基)吡啶-2-基]胺基}吡啶並[2,3-d]嘧啶-7(8H)-酮(即式Ⅰ所示的化合物)。II期臨床研究表明,palbociclib與乳腺癌的金標準治療藥物來曲唑相比,其治療活性提高3倍以上,無進展生存期(PFS)達26.1個月。palbociclib把以前對乳腺癌以「月」為標準的臨床治療效果提高到「年」的水平,取得了突破性的進展。正因為其良好的治療活性,FDA僅僅基於II期臨床數據,於2015年2月4日批准其在美國上市。目前報導的palbociclib晶型有WO2014128588A1公開的晶A和晶B,其中晶B不穩定,晶A為輝瑞的藥用晶型。但晶A易存在較嚴重的有機溶劑殘留,如苯甲醚和正丁醇這兩高沸點溶劑的殘留可達0.5wt%(WO2014128588A1,P17);在生產中我們也常發現苯甲醚和正丁醇殘留超過0.5wt%的情況,甚至可達1.0%左右。同時,晶A的製備需要異常嚴格的控溫條件,如需程序性降溫(WO2014128588A1,P43,P44),這非常不利於工業化大生產。此外,晶A結晶溶劑苯甲醚的生產中,必須使用劇毒性的硫酸二甲酯(孫寶國,香料與香精,中國石化出版社,2000:24),因而晶A的製備勢必導致嚴重的次級汙染問題。因此,本領域迫切需要提供一種理化性質穩定、有機溶劑殘留少、利於工業化生產、且製備過程無次級汙染問題的palbociclib新晶型。技術實現要素:本發明旨在提供一種化合物Ⅰ的穩定的新晶型。本發明的另一個目的是提供所述化合物Ⅰ的新晶型的製備方法。本發明的再一個目的是提供所述化合物Ⅰ的新晶型的用途。本發明的第四個目的是提供一種含有所述化合物Ⅰ的新晶型的藥物組合物。在本發明的第一方面,提供了一種化合物的晶型C,所述化合物結構如式Ⅰ所示,所述晶型C的X-射線粉末衍射(XRPD)有下述2θ角特徵峰:5.56±0.2°、7.68±0.2°和8.67±0.2°;;較佳地,所述晶型C的X-射線粉末衍射(XRPD)還有下述2θ角特徵峰:12.79±0.2°和16.36±0.2°;更佳地,所述晶型C的X-射線粉末衍射(XRPD)還有下述2θ角特徵峰:11.21±0.2°、18.14±0.2°、19.86±0.2°、21.24±0.2°、23.92±0.2°和26.58±0.2°;最佳地,所述晶型C有如圖1所示的X-射線粉末衍射(XRPD)圖。在另一優選例中,所述晶型C的差示掃描量熱法圖譜(DSC)上在25~300℃的溫度範圍內有一特徵峰,熔點為272.6℃;更佳地,所述晶型C有如圖2所示的差示掃描量熱法圖譜(DSC)。在另一優選例中,所述晶型C的紅外吸收圖譜在2949±2cm-1、1694±2cm-1、1654±2cm-1、1583±2cm-1和1400±2cm-1處有特徵峰;更佳地,所述晶型C有如圖4所示的紅外光譜。在另一優選例中,所述晶型C有一分子結晶水;較佳地,所述晶型C的TGA圖在50~275℃失重約3.88%;更佳地,所述晶型C有如圖3所示的TGA圖。在本發明的第二方面,提供了一種如上所述的本發明提供的式Ⅰ化合物的晶 型C的製備方法,所述的方法包括步驟:(1)將4-{6-[(6-(1-丁氧基乙烯基)-8-環戊基-5-甲基-7-氧代-7,8-二氫吡啶[2,3-d]嘧啶-2-基胺基]吡啶-3-基}哌嗪-1-甲酸叔丁酯與醇和水混合,得到溶液1,所述醇選自下述的一種或一種以上:正丁醇、甲醇、乙醇、異丙醇;(2)將溶液1和選自下述的一種或一種以上的酸混合,得到反應物2:鹽酸、硫酸、甲磺酸、羥乙基磺酸、三氟醋酸;(3)將反應物2冷卻至出現固體,過濾得到固體3;(4)將固體3與弱鹼水溶液混合,得到溶液4,所述弱鹼水溶液選自下述的一種或一種以上混合:飽和碳酸氫鈉、飽和碳酸氫鉀、飽和碳酸鈉、飽和碳酸鉀;和(5)將溶液4過濾,得到如上所述的本發明提供的式Ⅰ化合物的晶型C。在另一優選例中,在步驟(2)中使用鹽酸。在另一優選例中,步驟(2)中混合時間為4~24小時;更佳地,混合時間為4~8小時。在另一優選例中,步驟(3)中在0~4℃冷卻。在本發明的第三方面,提供了一種如上所述的本發明提供的式Ⅰ化合物的晶型C的用途,用於製備預防或治療乳腺癌、胃癌、肺癌、肝癌、甲狀腺癌、結腸直腸癌、神經膠質瘤、卵巢癌的藥物。在本發明的第四方面,提供了一種藥物組合物,所述的藥物組合物中含有如上所述的本發明提供的式Ⅰ化合物的晶型C和藥學上可接受的載體。據此,本發明提供了一種理化性質穩定、有機溶劑殘留少、利於工業化生產、且製備過程無次級汙染問題的palbociclib新晶型。附圖說明圖1顯示了本發明實施例4製備得到的化合物Ⅰ晶型C的X-射線粉末衍射圖;其中有關各個峰的2θ角如下表所示:#2-ThetaI%(相對峰強度)15.56544.227.678100.038.66639.5411.21111.1512.79140.5616.36358.6718.1414.9819.8577.4921.23917.81023.92416.21126.5878.8。圖2顯示了本發明實施例4製備得到的化合物I晶型C的差示掃描量熱法圖(DSC)。圖3顯示了本發明實施例4製備得到的化合物I晶型C的TGA圖譜。圖4顯示了本發明實施例4製備得到的化合物I晶型C的紅外光譜(IR)。圖5顯示了本發明實施例6製備得到的化合物Ⅰ晶型A和晶型C混晶的X-射線粉末衍射圖。圖6顯示了依據WO2014128588A1製備得到的化合物Ⅰ晶型A的X-射線粉末衍射圖。圖7顯示了依據WO2014128588A1製備得到的化合物Ⅰ晶型B的X-射線粉末衍射圖。具體實施方式發明人通過深入研究,發現化合物Ⅰ在醇和水中溶解後,通過酸水解獲得的固體通過弱鹼的水溶液處理,但無需使用苯甲醚等溶劑,便能獲得一種新的晶型,即單一晶型C,是一種穩定的晶型,且這種方法無需控溫,所得晶體沒有有機溶劑殘留。如本文所用,化學式或名稱應當包括所有的光學和立體異構體,以及存 在這些異構體和混合物的消旋混合物。化合物Ⅰ晶型C本發明提供了一種如式Ⅰ所示化合物的晶型C,並採用多種方式和儀器對其性質進行了研究。「X射線粉末衍射」,又稱「X射線多晶衍射(XRPD)」是目前用於測定晶型構造(即晶型)的常用試驗方法。採用X射線粉末衍射儀,在X射線透過晶型時產生一系列衍射圖譜,該圖譜中不同的衍射線及其強度由一定結構的原子團所決定,由此確定晶型的具體晶型結構。測定晶型的X射線粉末衍射的方法在本領域中是已知的。例如使用BrukerD8Advanced型號的X射線粉末衍射儀,以2°每分鐘的掃描速度,採用銅輻射靶獲取圖譜。本發明的化合物Ⅰ晶型C具有特定的晶型形態,在X-射線粉末衍射(XRPD)圖中具有特定的特徵峰。具體而言,本發明的化合物Ⅰ晶型C的X-射線粉末衍射(XRPD)圖上在下述2θ角有特徵峰:5.56±0.2°,7.68±0.2°和8.67±0.2°;較佳地,在下述2θ角還有特徵峰:12.79±0.2°和16.36±0.2°;更佳地,在下述2θ角還有特徵峰:11.21±0.2°、18.14±0.2°、19.86±0.2°、21.24±0.2°、23.92±0.2°和26.58±0.2°。最佳地,所述具有與圖1基本一致的X-射線粉末衍射(XRPD)圖。「示差掃描量熱分析」,又稱「差示量熱掃描分析」(DSC)是在加熱過程中,測量被測物質與參比物之間的能量差與溫度之間關係的一種技術。DSC圖譜上的峰位置、形狀和峰數目與物質的性質有關,故可以定性地用來鑑定物質。本領域常用該方法來檢測物質的相變溫度、玻璃化轉變溫度、反應熱等多種參 數。DSC測定方法在本領域中是已知的。例如可使用DSCQ20示差掃描量熱分析儀,以10℃每分鐘的升溫速率,從25℃升溫至300℃,獲得晶型的DSC掃描圖譜。在本發明的一個實施方式中,採用DSC測得用本發明方法獲得的化合物Ⅰ晶型C在25~300℃範圍內有一特徵峰,熔點為272.6℃;更優選本發明的化合物I晶型C具有與圖2基本一致的DSC圖譜。熱重分析(ThermogravimetricAnalysis,TG或TGA)在本領域中是已知的。在程序控制溫度下測量樣品的質量與溫度變化關係。在本發明的一個實施方式中,採用TGA測得用本發明方法獲得的化合物Ⅰ晶型C有一分子結晶水;優選在50~275℃失重約3.88%;更優選本發明的化合物I晶型C具有與圖3基本一致的TGA圖譜。也可採用紅外圖譜法(IR)來確定晶型結構,其測定方法在本領域中是已知的。例如可採用PESpectrumOneB,以KBr:樣品=200:1壓片,並在400-4000cm-1範圍掃描。本發明化合物Ⅰ的晶型C的紅外圖譜顯示以下波數有特徵峰:2949±2cm-1、1694±2cm-1、1654±2cm-1、1583cm±2-1和1400±2cm-1,優選具有與圖4基本一致的紅外圖譜。化合物Ⅰ晶型C的製備方法本發明還提供了式Ⅰ所示化合物的晶型C的製備方法。在本發明提供的一種實施方式中,式Ⅰ化合物的晶型C的製備方法包括以下步驟:第一步,將4-{6-[(6-(1-丁氧基乙烯基)-8-環戊基-5-甲基-7-氧代-7,8-二氫吡啶[2,3-d]嘧啶-2-基胺基]吡啶-3-基}哌嗪-1-甲酸叔丁酯(cas:866084-31-3)與醇和水混合,得到溶液1,所述醇選自下述的一種或一種以上:正丁醇、甲醇、乙醇、異丙醇;第二步,將溶液1和選自下述的一種或一種以上的酸混合,得到反應物2:鹽酸、硫酸、甲磺酸、羥乙基磺酸、三氟醋酸;第三步,將反應物2冷卻至出現固體,過濾得到固體3;第四步,將固體3與弱鹼水溶液混合,得到溶液4,所述弱鹼水溶液選自下述的一種或一種以上混合:飽和碳酸氫鈉、飽和碳酸氫鉀、飽和碳酸鈉、飽和碳酸鉀;第五步,將溶液4過濾,得到本發明提供的化合物Ⅰ的晶型C。在上述第一步中,所述混合在加熱條件下進行,例如但不限於,在60-80℃下加熱20-40分鐘。在上述第二步中使用的酸優選鹽酸。在上述第二步中,所述混合是水解反應,優選在加熱條件下進行,例如但不限於,在60~80℃下反應4-24小時;優選4~8小時。在上述第三步中,可以使用本領域常規的方法對反應物2進行冷卻,例如但不限於,在0~4℃冰箱過夜,使大量固體出現。在上述第三步中,過濾後用醇洗滌固體,所使用的醇一般與第一步中的相同;在本發明的一個具體實施例中,洗滌後置於45~55℃空氣浴中乾燥,得到固體3,所述固體3是palbociclib的鹽酸鹽、羥乙基磺酸鹽等固體。在上述第四步中,所述混合在攪拌下進行,在本發明的一個具體實施例中,在15~30℃攪拌2~12小時。在上述第五步中,在本發明的一個具體實施例中,過濾後得到的固體用蒸餾水洗滌,洗滌後置於45~55℃空氣浴中乾燥,得到本發明提供的化合物Ⅰ的晶型C。化合物Ⅰ晶型C的用途本發明提供的式Ⅰ化合物的晶型C穩定性好,可以作為原料藥,例如用於製備預防或治療乳腺癌、胃癌、肺癌、肝癌、甲狀腺癌、結腸直腸癌、神經膠質瘤、卵巢癌的藥物。所述的藥物包括式Ⅰ化合物的晶型C和藥學上可接受的載體。所述藥物可以根據不同給藥途徑而製備成各種劑型。這些劑型以下面方式之一施用:膠囊、片劑、口崩片、含片、緩控釋製劑等。另外需要指出,本發明式Ⅰ化合物的晶型C的使用劑量和使用方法取決於諸多因素,包括患者的年齡、體重、性別、自然健康狀況、營養狀況、化合物活性強度、服用、代謝速率、病症的嚴重程度以及診治醫師的主觀判斷。定義如本文所用,「如式Ⅰ所示的化合物」、「式Ⅰ化合物」或「化合物Ⅰ」可以互換使用,都是指無定形態的結構式如Ⅰ所示的化合物;優選使用WO2014128588A1公開的方法製備得到的4-{6-[(6-(1-丁氧基乙烯基)-8-環戊基 -5-甲基-7-氧代-7,8-二氫吡啶[2,3-d]嘧啶-2-基胺基]吡啶-3-基}哌嗪-1-甲酸叔丁酯。如本文所用,「本發明palbociclib的晶型C」、「晶型C」或「化合物Ⅰ的晶型C」可互換使用,都是指X-射線粉末衍射(XRPD)圖上在下述2θ角有特徵峰:5.56±0.2°、7.68±0.2°和8.67±0.2°;優選在下述2θ角還有特徵峰:12.79±0.2°和16.36±0.2°;更優選在下述2θ角還有特徵峰:11.21±0.2°、18.14±0.2°、19.86±0.2°、21.24±0.2°、23.92±0.2°和26.58±0.2°。如本文所用,術語「藥學上可接受的載體」指用於治療劑給藥的載體,包括各種賦形劑和稀釋劑。該術語指這樣一些藥劑載體:它們本身並不是必要的活性成分,且施用後沒有過分的毒性。合適的載體是本領域普通技術人員所熟知的。在Remington'sPharmaceuticalSciences(MackPub.Co.,N.J.1991)中可找到關於藥學上可接受的賦形劑的充分討論。在組合物中藥學上可接受的載體可包括液體,如水、鹽水、甘油和乙醇。另外,這些載體中還可能存在輔助性的物質,如崩解劑、潤溼劑、乳化劑、pH緩衝物質等。本發明提到的上述特徵,或實施例提到的特徵可以任意組合。本案說明書所揭示的所有特徵可與任何組合物形式並用,說明書中所揭示的各個特徵,可以任何可提供相同、均等或相似目的的替代性特徵取代。因此除有特別說明,所揭示的特徵僅為均等或相似特徵的一般性例子。本發明的主要優點在於:1、本發明提供的式Ⅰ化合物的晶型C具有良好的化學穩定性和晶型穩定性。2、本發明提供的式Ⅰ化合物的晶型C沒有有機溶劑殘留,並且重金屬鈀的含量很低。3、本發明提供的式Ⅰ化合物的晶型C的藥代性質與晶型A藥代性質相當。4、本發明提供的式Ⅰ化合物的晶型C的製備方法操作簡單,適合工業化生產。5、本發明提供的式Ⅰ化合物的晶型C的製備方法中無次級汙染,綠色環保。下面結合具體實施例,進一步闡述本發明。應理解,這些實施例僅用於說明本發明而不用於限制本發明的範圍。下列實施例中未註明具體條件的實驗方法,通常按照常規條件或按照製造廠商所建議的條件。除非另外說明,否則所有的百分數、比率、比例、或份數按重量計。本發明中的重量體積百分比中的單位是本領域技術人員所熟知的,例如是指在100毫升的溶液中溶質的重量。除非另行定義,文中所使用的所有專業與科學用語與本領域熟練人員所熟悉的意義相同。此外,任何與所記載內容相似或均等的方法及材料皆可應用於本發明方法中。文中所述的較佳實施方法與材料僅作示範之用。原料和通用測試方法實施例中所用4-{6-[(6-(1-丁氧基乙烯基)-8-環戊基-5-甲基-7-氧代-7,8-二氫吡啶[2,3-d]嘧啶-2-基胺基]吡啶-3-基}哌嗪-1-甲酸叔丁酯(cas:866084-31-3)為參考WO2014128588A1所製備。核磁共振波譜儀:VarianMercury400NMRspectrometerX-射線粉末衍射儀器:BruckerD8advanceX-射線粉末衍射儀;輻射源:銅靶在室溫條件下掃描;電壓:40kv;電流:40mA;起始的2θ:2.000°,掃描範圍:2.0000-50.000°;步長:0.020°;測量時間:12.6秒/步。差示掃描量熱法分析(DSC)儀器:美國TA公司的Q2000型,20-200℃範圍內,加熱速率10℃/min,氮氣流速50ml/min。熱重分析(TGA)儀器:美國TA公司的SDTQ600型,20-300℃範圍內,加熱速率10℃/min,氮氣流速60ml/min。紅外分光光度法(FTIR)分析儀器:Nicolet380傅立葉變換紅外光譜儀;溴化鉀壓片法;解析度:4.0cm-1。電感耦合等離子光譜儀:Optima5300DV。實施例1palbociclib鹽酸鹽的製備將4-{6-[(6-(1-丁氧基乙烯基)-8-環戊基-5-甲基-7-氧代-7,8-二氫吡啶[2,3-d]嘧啶-2-基胺基]吡啶-3-基}哌嗪-1-甲酸叔丁酯(31.5g,52.2mmoL)加入到正丁醇(548.0mL)和蒸餾水(315.0mL)的混合溶液中,於油浴70℃加熱30分鐘。加入濃鹽酸(22.0mL),在油浴70℃反應過夜。冷卻,反應體系放置0℃冰箱過夜,出現大量固體。過濾,固體用正丁醇(5.0mL)洗滌,於50℃空氣浴中乾燥至恆重,得純黃色固體25.0g。1HNMR(400MHz,D2O):δ=8.94(s,1H),8.00(dd,J1=2.1Hz,J2=9.6Hz,1H),7.76(d,J1=2.0Hz,1H),7.41(d,J1=2.1Hz,J2=9.2Hz,1H),5.72(m,1H),3.48(dd,J1=4.0Hz,J2=7.2Hz,4H),3.41(dd,J1=2.4Hz,J2=6.0Hz,4H),2.43(s,3H),2.31(s,3H),2.08(m,2H),1.93(m,4H),1.64(m,2H).實施例2palbociclib甲磺酸鹽的製備將4-{6-[(6-(1-丁氧基乙烯基)-8-環戊基-5-甲基-7-氧代-7,8-二氫吡啶[2,3-d]嘧啶-2-基胺基]吡啶-3-基}哌嗪-1-甲酸叔丁酯(3.15g,5.22mmoL)加入到正丁醇(55.0mL)和蒸餾水(31.50mL)的混合溶液中,於油浴70℃加熱30分鐘。加入甲磺酸(2.0mL),在油浴70℃反應過夜。冷卻,反應體系放置0℃冰箱過夜,出現大量固體。過濾,固體用正丁醇(5.0mL)洗滌,於50℃空氣浴中乾燥至恆重,得純黃色固體3.0g。實施例3palbociclib羥乙基磺酸鹽的製備將4-{6-[(6-(1-丁氧基乙烯基)-8-環戊基-5-甲基-7-氧代-7,8-二氫吡啶[2,3-d]嘧啶-2-基胺基]吡啶-3-基}哌嗪-1-甲酸叔丁酯(7.25g,12.0mmoL)加入到甲醇(140mL)中,再緩慢滴加羥乙基磺酸(5.3g,42.0mmoL)、甲醇(15.0mL)和水(0.70mL,38.9mmoL)的混合溶液。滴加完畢後,體系加熱至55℃,且在此溫度下攪拌30分鐘。滴加三乙胺(1.8g,13.0mmoL)的甲醇(3.0mL)溶液,在此過程中反應體系溫度降至30℃。在30℃下滴加三乙胺(1.3g,12.6mmoL)的甲醇(20.0mL),6.0小時滴加完畢。攪拌過夜。於4℃下放置1.0小時,過濾,固體用並甲醇(2.0mL)洗滌,於55℃真空乾燥1.0小時,得純黃色固體6.8g,收率為87%。1HNMR(400MHz,DMSO-d6:δ=10.23(s,1H),8.98(s,1H),8.69(brs,1H),8.12(d,J=3.2Hz,1H),7.92(d,J=9.2Hz,1H),7.55(dd,J1=2.8Hz,J2=8.8Hz,1H),5.84(m,1H),4.46(brs1H),3.62(t,J=6.8Hz,2H),3.37(m,4H),3.27(m,4H),2.60(t,J=6.8Hz,2H),2.43(s,3H),2.32(s,3H),2.23(m,2H),1.93(m,2H),1.88(m,2H),1.63(m,2H).實施例4palbociclib晶型C的製備將實施例1所得產物(1.0g)緩慢加入到碳酸氫鈉飽和水溶液(12.0mL)和蒸餾水(15.0mL)中,於室溫攪拌4.0小時。過濾,固體用蒸餾水(3×5.0mL)洗滌,於50℃空氣浴中乾燥至恆重,得0.72g晶型C。1HNMR(400MHz, DMSO-d6+TFA):δ=11.18(brs,1H),9.01(s,3H),8.07(d,J=2.8Hz,1H),7.95(dd,J1=2.8Hz,J2=9.6Hz,1H),7.81(d,J=9.2Hz,1H),5.85(m,1H),3.43(t,J=4.8Hz,4H),3.30(s,4H),2.44(s,3H),2.35(s,3H),2.22(m,2H),1.94(m,2H),1.93(m,2H),1.60(m,2H).13CNMR(100MHz,DMSO-d6+TFA):δ=202.1,160.51,158.97,158.60,158.23,157.86,157.32,157.02,154.97,143.36,142.14,141.51,130.92,130.85,119.73,116.85,116.36,113.96,111.08,53.11,45.19,42.39,31.16,27.61,25.19,13.67.HRMS(ESI+,m/z):448.2453[M+H]+.結果對所得晶型C進行XRPD測試、DSC測試、TGA測試、FTIR測試:從圖1中可以看出,在2θ值為5.56±0.2°、7.68±0.2°、8.67±0.2°、12.79±0.2°、16.36±0.2°處具有強峰;在11.21±0.2°、18.14±0.2°、19.86±0.2°、21.24±0.2°、23.92±0.2°、26.58±0.2°處具有次強峰。從圖2中可以看出,在25-200℃範圍內有單一吸熱峰,onset約272.6℃。從圖3中可以看出,在50~275℃範圍內失重約3.88%,表示含有1分子結晶水。圖4是本實施例製備的晶型C的FTIR圖譜,從圖4中可以看出,在2949±2cm-1、1694±2cm-1、1654±2cm-1、1583±2cm-1、1400±2cm-1等處有特徵峰。實施例5palbociclib晶型C的製備將實施例1所得產物(1.0g)緩慢加入到碳酸氫鉀飽和溶液(12.0mL)和蒸餾水(15.0mL)中,於室溫攪拌4.0小時。過濾,固體用蒸餾水(3×5.0mL)洗滌,於50℃空氣浴中乾燥至恆重,得0.73g晶型C。結果所得晶型C的XRPD圖譜、DSC圖譜、TGA圖譜、FTIR圖譜基本同實施例4一致。實施例6palbociclib晶型A和晶型C混晶的製備將實施例3所得產物(0.72g)緩慢加入到碳酸氫鈉飽和溶液(13.0mL)和蒸餾水(15.0mL)中,於室溫攪拌4.0小時。過濾,固體用蒸餾水(3×5.0mL)洗滌,於50℃空氣浴中乾燥至恆重,得0.68g純黃色固體。結果所得固體的XRPD圖譜表明,此黃色固體為晶A和晶C的混晶。此混晶的XRPD如圖5所示。實施例7palbociclib晶型C的溶劑殘留研究依次採用HPLC法和GC法測量了晶型C的可能溶劑苯甲醚、正丁醇和正庚烷的殘留,結果如表1所示。晶型C中未檢出有苯甲醚、正丁醇和正庚烷。表1晶型C的可能溶劑苯甲醚、正丁醇和正庚烷的殘留研究溶劑檢測方法檢測結果苯甲醚HPLC未檢出正丁醇GC未檢出正庚烷GC未檢出實施例8palbociclib晶型C的重金屬元素鈀的含量研究採用EPA6010C電感耦合等離子體原子發射光譜法檢測了晶型C中的重金屬元素鈀的含量,結果如表2所示。晶型C中的重金屬鈀含量很低,小於1.0ppm。表2晶型C的的重金屬元素鈀的含量研究樣品編號檢測項目檢測結果(mg/kg)2015042801Pd<1實施例9palbociclib晶型C的藥代研究在大鼠體內研究了晶型C和晶型A的藥代性質。藥代試驗方法SD大鼠,180~220g。靜脈注射給藥溶液的配製:先將palbociclib晶型C和晶型A分別配成100mM的DMSO溶液;再分別取palbociclib晶型C和晶型A的100mM母液各為19.0μL,稀釋至1.7mL(N.S:PEG400=7:3,含20μLTween80),超聲3min,配製為0.5mg/mL的溶液。灌胃給藥混懸液配製:分別稱取palbociclib晶型C和晶型A化合物各2.72mg,加入7.30mL的CMC-Na(0.5%),配製為0.375mg/mL的混懸液體(不溶)。palbociclib晶型C給操作:SD大鼠5隻,2隻靜脈注射給予5.0mg/kg的晶型C,分別於給藥前和給藥後2、5、15、30、60、90、120、240、360min於大鼠眼底靜脈叢取血0.4mL。3隻灌胃給予5.0mg/kg的晶型C,分別於給藥前和給藥後5、15、30、60、90、120、240、360min於大鼠眼底靜脈叢取血0.4mL。血樣於8000rpm離心5min,取血漿於離心管中4℃保存備用。血漿樣品50μL,加入200μL含內標的乙腈(R822.5uM)沉澱蛋白,渦旋10min,6000g離心10min,取200μL上清6000g再次離心10min,取上清70μL於96孔板中進樣,進樣量5μL。靜脈注射前4個時間點,用空白血漿稀釋10倍後測定。palbociclib晶型A給藥操作:操作同晶型C。試驗結果如表3所示。表3Palbociclib晶型C和晶型A的藥代研究此研究表明,palbociclib晶型C和晶型A的藥代性質相當,二者無顯著性差異。實施例10palbociclib晶型C的穩定性研究取實施例4所製備得到晶型C分別放置在不同溫度、溼度條件下,考察其化學穩定性和晶型穩定性,結果如表4所示。表4Palbociclib晶型C的穩定性研究由上述實驗可以看出,本發明製備得到的晶型C具有良好的化學穩定性和晶型穩定性,在室溫及較高溫度下如60℃下均能夠長時間穩定保存。以上所述僅為本發明的較佳實施例而已,並非用以限定本發明的實質技術內容範圍,本發明的實質技術內容是廣義地定義於申請的權利要求範圍中,任何他人完成的技術實體或方法,若是與申請的權利要求範圍所定義的完全相 同,也或是一種等效的變更,均將被視為涵蓋於該權利要求範圍之中。當前第1頁1 2 3