一種高純度依達拉奉的合成工藝的製作方法
2023-06-05 14:01:56 1
本發明涉及一種已知化合物的合成方法,具體地說是一種高純度依達拉奉的合成工藝,屬於醫藥
技術領域:
。二、
背景技術:
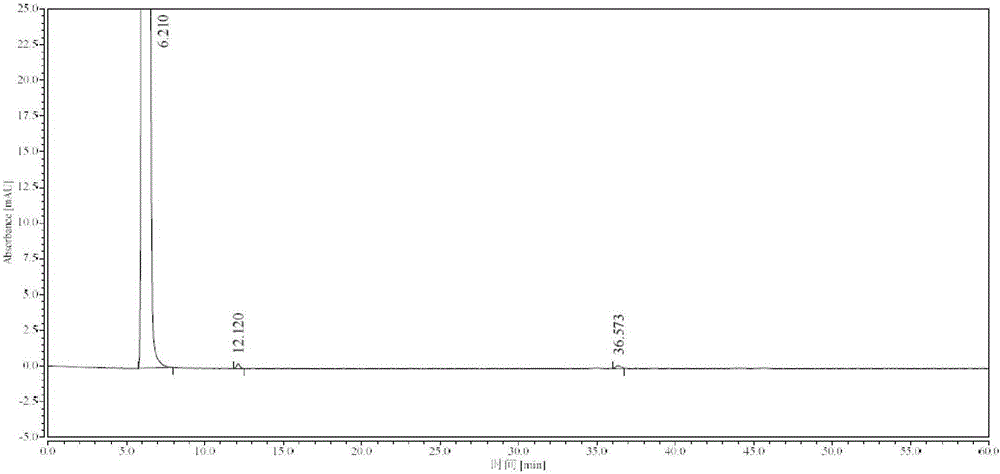
:依達拉奉(Edaravone,式(I)),化學名為3-甲基-1-苯基-2-吡唑啉-5-酮,由日本三菱化學株式會社開發,2001年6月在日本上市。依達拉奉是一種自由基清除劑,具有很強的自由基清除及抗過氧化作用,臨床用於改善急性腦梗塞所致的神經症狀、日常生活活動能力和功能障礙。依達拉奉是臨床上第一個用於治療急性腦梗死的自由基清除劑,並且是第一個不影響纖溶系統的缺血神經元保護劑,具有較高的臨床應用價值和廣闊的市場前景。藥用依達拉奉的製備一般採用苯肼與乙醯乙酸乙酯合成法,可用乙醇、水或醋酸為溶劑,在酸性環境(加酒石酸、鹽酸、硫酸、亞硫酸氫鈉等)或鹼性環境(不加任何催化劑或輔助劑)下加熱回流反應2-3h,冷卻析晶,即得依達拉奉。此方法所用原料廉價易得,反應操作簡單,是目前較通用的方法,合成路線如下:此反應的機理為:乙醯乙酸乙酯與苯肼先經伯胺加成縮合再經分子內酯氨解(仲胺)環合得依達拉奉。反應機理如下:由以上機理可知:乙醯乙酸乙酯分子中含有兩種羰基結構:酮羰基和酯羰基;苯肼分子中也含有兩種氨基結構:伯胺和仲胺基。由此,乙醯乙酸乙酯與苯肼可發生兩類反應:席夫鹼反應(伯胺與酮縮合)、酯氨解反應,均通過親核加成-消除機理進行反應;從空間位阻看最容易發生的是席夫鹼反應,其次是酯氨解反應;席夫鹼反應有水生成,而乙醯乙酸乙酯會水解,無水環境對反應更為有利;苯肼伯胺反應活性強,在高溫或過量投料情況下,會與乙醯乙酸乙酯同時發生席夫鹼反應和酯氨解反應,產生雙縮合雜質;依達拉奉為自由基清除劑,易氧化變色,反應溫度升高或反應時間延長均會加劇依達拉奉氧化雜質生成,使成品帶色、純度降低。因此,苯肼不宜過量投料,且投料過程中為防止局部濃度過高,也不能與乙醯乙酸乙酯同時投料,要乙醯乙酸乙酯先投,苯肼後下,苯肼宜在低溫慢速投料,低溫快速反應有利於改善產品顏色和純度。專利CN201110282520.2以過量的苯肼或者鹽酸苯肼投料,產生雙縮合雜質不可避免,且工藝中用到大量連二硫酸鈉作耗氧劑和催化劑,危險性高,僅適合實驗室少量合成,不適於工業生產;專利US4857542在乙醇中回流反應3h,依達拉奉氧化降解明顯,精製除雜代價大,精製後收率僅65%。現有技術合成依達拉奉均需要在酸性環境或不加催化劑條件下高溫(大於75℃)反應,反應時間不少於2h,產品顏色深,雜質多,僅適合實驗室少量合成,工業化生產需要反覆精製,產品顏色、雜質才能達到藥用要求,實際收率低,成本較高。三、技術實現要素:本發明旨在提供一種高純度依達拉奉的合成工藝,本方法操作簡便、成本低、收率高,產品顏色及純度符合注射用依達拉奉原料藥要求。本發明在對依達拉奉的合成工藝條件進行篩選過程中,發現影響依達拉奉顏色的主要參數為苯肼投料溫度,當投料溫度大於30℃時,合成的依達拉奉顏色明顯加深,發明人試圖在精製溶液中加活性炭、硅藻土、氧化鋁等吸附劑脫色,結果加活性炭有炭溶現象,硅藻土和氧化鋁均不脫色。進一步研究發現酯氨解環合反應在適量鹼(氫氧化鈉、氫氧化鉀、氫化鈉等)的醇(無水乙醇、異丙醇)溶液中更容易發生,75-85℃回流反應30min即可反應完全,但產品顏色隨鹼量的增加而加深,雜質也會增多,經試驗,當鹼與苯肼的摩爾比值大於0.009時,產品顏色加深明顯,而其摩爾比值小於0.005時,對反應時間沒有明顯影響,當其摩爾比值在0.007-0.009,可以獲得反應時間較短、收率高的產品,並且產品顏色和純度均符合藥用要求。因此,本發明包含苯肼低溫投料、加鹼催化環合過程。本發明高純度依達拉奉的合成工藝,包括如下步驟:將乙醯乙酸乙酯溶於低級醇中,在15-25℃下滴加苯肼,滴加完畢後於15-25℃下攪拌10min,隨後加入催化量的鹼,加熱至回流反應;反應完成後降溫攪拌析晶,過濾,濾餅用乙酸乙酯洗滌至濾液無色,再用無水乙醇重結晶即得目標產物——白色結晶依達拉奉(式I)。所述低級醇選自無水甲醇、乙醇、無水乙醇、異丙醇中的一種或一種以上的混合物,優選為乙醇、無水乙醇或異丙醇,更優選為無水乙醇或異丙醇。所述乙醯乙酸乙酯與苯肼的摩爾比為1:0.95-1.0。所述鹼選自甲醇鈉、乙醇鈉、叔丁醇鈉、叔丁醇鉀、氫氧化鈉、氫氧化鉀中的一種或一種以上的混合物,優選為氫氧化鈉或氫氧化鉀,更優選為氫氧化鈉。所述乙醯乙酸乙酯與鹼的摩爾比為1:0.007-0.009。加熱至回流反應的反應溫度為75-85℃,反應時間為0.5-1.5h。攪拌析晶過程控制析晶溫度為0-10℃,析晶時間為1-3h。重結晶時濾餅與無水乙醇的質量體積比為1g:2-3mL。重結晶的溫度控制在0-10℃,時間為1-3h。反應過程如下:本發明製得的依達拉奉為白色結晶。本發明製得的依達拉奉採用雜質對照法檢測,已知苯肼、乙醯乙酸乙酯、二聚體及三聚體均未檢出,純度大於99.97%(HPLC法),結果見表1,色譜圖見圖1-3。分析方法如下:色譜柱:AcclaimTM120C18(250mm×4.6mm,5.0μm)流動相:0.05mol/L磷酸二氫銨溶液(用20%磷酸溶液調節pH值至3.5)-甲醇(50︰50)檢測波長:245nm溶劑:流動相樣品濃度:1mg/ml流速:1.0ml/min進樣量:10μl表1依達拉奉有關物質檢測結果名稱相對保留時間無水乙醇結晶前無水乙醇結晶後苯肼0.55未檢出未檢出乙醯乙酸乙酯0.71未檢出未檢出二聚體1.960.019%未檢出三聚體5.940.011%未檢出其他最大單雜/未檢出未檢出總雜/0.030%0本發明製得的依達拉奉採用氣相色譜法檢測殘留溶劑含量,結果溶劑殘留量均符合現行藥典規定。另外,苯肼及其可能含有的雜質苯胺、聯苯胺均為基因毒性雜質,經HPLC外標對照法檢測本發明製得的依達拉奉中的苯肼、苯胺及聯苯胺含量,結果均未檢出,質量安全性可靠。與現有的技術相比,本發明的有益效果是:本發明通過控制苯肼投料溫度、投料量和投料順序,從源頭控制有色雜質的產生;加鹼催化環合反應,縮短反應時間,減少雜質生成時間,提高產品純度,節約了生產成本,產品顏色符合注射用依達拉奉原料藥要求,產品純度大於99.97%(HPLC法)。四、附圖說明圖1是依達拉奉雜質定位HPLC色譜圖。圖2是依達拉奉無水乙醇結晶前HPLC色譜圖。圖3是依達拉奉無水乙醇結晶後HPLC色譜圖。五、具體實施方式下面對本發明的技術方案進行說明,以便於本
技術領域:
的技術人員理解。實施例1:取乙醯乙酸乙酯18kg(138.31mol),加無水乙醇21L溶解,控溫15-25℃,滴加苯肼14.9kg(137.78mol),滴畢,於15-25℃保溫攪拌10min,加入氫氧化鈉44g(1.1mol),80℃回流反應1h,攪拌降溫至0-10℃,攪拌析晶1h,過濾,濾餅用乙酸乙酯洗至濾液無色,得依達拉奉溼品23kg,濾餅用57L無水乙醇回流溶解,過濾,濾液攪拌降溫至0-10℃,攪拌析晶3h,過濾,用4L冷無水乙醇洗滌濾餅,60-70℃真空乾燥(≥0.09MPa)6h,得白色結晶依達拉奉17.1kg,總收率71.2%,純度大於99.97%(HPLC法)。實施例2:取乙醯乙酸乙酯18kg(138.31mol),加異丙醇21L溶解,控溫15-25℃,滴加苯肼14.9kg(137.78mol),滴畢,於15-25℃保溫攪拌10min,加入氫氧化鈉44g(1.1mol),85℃回流反應45min,攪拌降溫至0-10℃,攪拌析晶1h,過濾,濾餅用乙酸乙酯洗至濾液無色,得依達拉奉溼品23.5kg,濾餅用58L無水乙醇回流溶解,過濾,濾液攪拌降溫至0-10℃,攪拌析晶3h,過濾,用4L冷無水乙醇洗滌濾餅,60-70℃真空乾燥(≥0.09MPa)6h,得白色結晶依達拉奉16.8kg,總收率70.0%,純度大於99.97%(HPLC法)。實施例3:取乙醯乙酸乙酯18kg(138.31mol),加無水乙醇21L溶解,控溫15-25℃,滴加苯肼14.9kg(137.78mol),滴畢,於15-25℃保溫攪拌10min,加入氫氧化鉀62g(1.1mol),76℃回流反應1.5h,攪拌降溫至0-10℃,攪拌析晶1h,過濾,濾餅用乙酸乙酯洗至濾液無色,得依達拉奉溼品23.3kg,濾餅用58L無水乙醇回流溶解,過濾,濾液攪拌降溫至0-10℃,攪拌析晶3h,過濾,用4L冷無水乙醇洗滌濾餅,60-70℃真空乾燥(≥0.09MPa)6h,得白色結晶依達拉奉17.0kg,總收率70.8%,純度大於99.97%(HPLC法)。實施例4:取乙醯乙酸乙酯180kg(1383.1mol),加無水乙醇210L溶解,控溫15-25℃,滴加苯肼149kg(1377.8mol),滴畢,於15-25℃保溫攪拌10min,加入氫氧化鈉440g(11mol),80℃回流反應1.5h,攪拌降溫至0-10℃,攪拌析晶1.5h,過濾,濾餅用乙酸乙酯洗至濾液無色,得依達拉奉溼品238kg,濾餅用590L無水乙醇回流溶解,過濾,濾液攪拌降溫至0-10℃,攪拌析晶3h,過濾,用40L冷無水乙醇洗滌濾餅,60-70℃真空乾燥(≥0.09MPa)6h,得白色結晶依達拉奉175kg,總收率72.9%,純度大於99.97%(HPLC法)。當前第1頁1 2 3