一種殼聚糖填充微型基質固相分散方法與流程
2023-04-25 02:08:16 4
本發明屬於天然藥物提取領域,涉及一種提取測定青果中有效成分的方法,具體涉及一種採用殼聚糖作為吸附劑的基質固相分散技術提取檢測青果中有效成分的方法。
背景技術:
青果為橄欖科植物橄欖Canarium album Raeusch.的乾燥成熟果實。秋季果實成熟時採收,乾燥。具有清熱解毒,利咽,生津等功能。可用於咽喉腫痛,咳嗽痰黏,煩熱口渴,魚蟹中毒等。青果一般用於鮮吃、加工涼果和製藥。不同品種的青果某些化學成分的含量差別很大,而對不同用途的青果的藥效成分有不同的要求,因此應對青果不同品種中的有效成分進行分析測定。在分析檢測之前需要對樣品進行預處理,傳統的預處理過程(例如超聲提取,回流提取,索氏提取等)一般包括提取和淨化兩步,因而它存在操作複雜、耗時、有機溶劑消耗量大,汙染環境等缺點。
基質固相分散(MSPD)是一種樣品前處理的新技術,由於它集樣品分散、提取和淨化與一體,因此相比於傳統的方法可簡化步驟,節約時間。目前傳統的基質固相分散方法的吸附劑量(1~10g)、藥材的用量(1g以上)、洗脫劑的用量(2~20ml)都比較大,其所用的吸附劑一般為氧化鋁,C18,矽膠等,他們普遍缺乏選擇性。因此開發小型的新型的基質固相分散方法也是非常有意義的。
殼聚糖是甲殼素脫乙醯基產物,它具有游離的氨基和羥基,有良好的吸附性能,殼聚糖及其衍生物具有吸附容量大、成本低、可再生性、有生物相容性、無毒、易於化學改性等優點,因而被廣泛應用於醫藥、紡織、水處理、生物工程等領域。由於殼聚糖具有良好的吸附性能,安全無毒,因此適合用作基質固相分散的吸附劑。
目前將殼聚糖作為吸附劑結合微型基質固相分散技術用於青果中化學成分的測定尚未見報導。本發明採用殼聚糖作為基質固相的吸附劑,並極大地減少了傳統基質固相萃取技術的樣品用量,並可高效提取青果的有效成分。
技術實現要素:
本發明是要解決目前提取檢測青果中有效成分的方法中步驟複雜、有機溶劑 用量大、費時、選擇性低等問題,採用殼聚糖為吸附劑的基質固相分散技術提取檢測青果中的有效成分的方法。
殼聚糖作為吸附劑的基質固相分散技術提取檢測青果中的有效成分的方法,具體是按照以下步驟完成的:
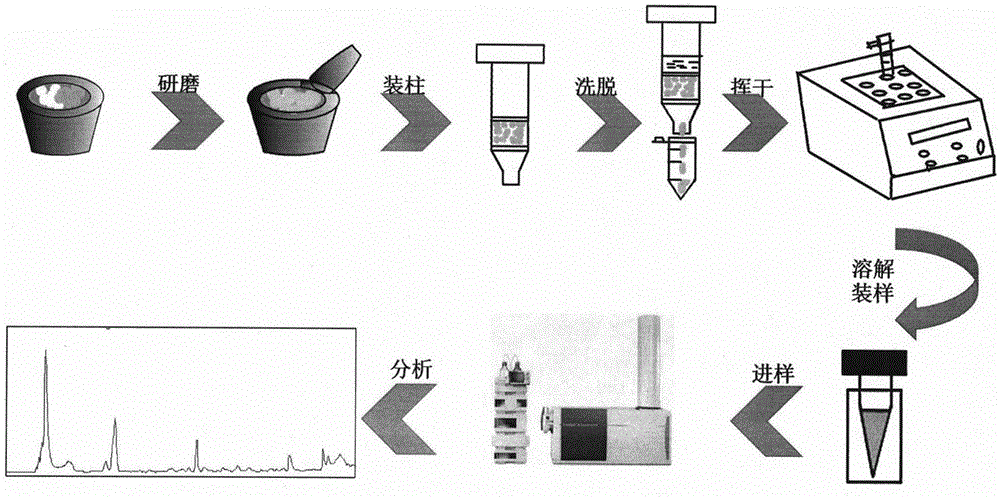
(1)稱取青果粉末50mg與一定質量比的吸附劑於研缽中,研磨一定的時間,將研磨好的固體作為填料裝入1ml規格的固相萃取小柱中,用0.5ml的洗脫液對小柱洗脫三次,將收集到的洗脫液揮幹,再用200μl的洗脫液溶解,將離心管內的液體渦旋10s後13000rpm離心5min。取上層清液裝樣,用超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)採樣分析,得到青果有效成分的色譜圖。
(2)將沒食子酸對照品配製成不同濃度的對照品溶液,按富集樣液的條件用超高效液相色譜-四極杆飛行時間質譜聯用儀檢測,獲得沒食子酸的譜圖,以沒食子酸的進樣量為橫坐標,以沒食子酸對照品溶液的譜圖中的峰面積為縱坐標製作沒食子酸標準曲線,按同樣方法製作羥基酪醇、沒食子酸甲酯、木犀草苷、蘆丁、橄欖苦苷、鞣花酸的標準曲線。根據提取液各成分色譜圖中的峰面積和各成分的標準曲線,計算得到提取液中各成分的含量。
其中(1)中所述吸附劑包括:中分子殼聚糖、低分子殼聚糖、低粘度殼聚糖、高純度殼聚糖、羧化殼聚糖,優選為中分子殼聚糖。
所述吸附劑的質量為:6.25mg、12.5mg、25mg、37.5mg、50mg、62.5mg、75mg、87.5mg,優選為25mg。
所述研磨時間為:20s、40s、60s、80s、100s,優選為60s。
所述洗脫液包括:60%甲醇、80%甲醇、100%甲醇、乙腈、乙醇、乙酸乙酯、正丁烷,優選為60%甲醇。
本發明的有益效果是:本發明採用基質固相分散技術提取青果中的有效成分,是提取和淨化一步完成,具有操作簡便,節約時間,有機溶劑及樣品用量少等優點。其選用殼聚糖作為吸附劑具有吸附性能好,成本低,安全無毒,可再生性等特點,提高了分析方法的選擇性。使其大大優於傳統的提取方法。此外本發明以超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)作為檢測器具有快速、穩定、準確的特點。
附圖說明
圖1為本發明基質固相分散萃取方法的工藝流程圖。
圖2為考察吸附劑種類的基質固相分散萃取效果柱狀圖。圖中,1、2、3、4、5、6、7分別代表沒食子酸、羥基酪醇、沒食子酸甲酯、木犀草苷、蘆丁、鞣花酸、橄欖苦苷。
圖3為考察藥材和中分子殼聚糖之間不同質量比例下的基質固相分散萃取效果折線圖。圖中,1、2、3、4、5、6、7分別代表沒食子酸、羥基酪醇、沒食子酸甲酯、木犀草苷、蘆丁、鞣花酸、橄欖苦苷。
圖4為考察不同研磨時間的基質固相分散萃取效果折線圖。圖中,1、2、3、4、5、6、7分別代表沒食子酸、羥基酪醇、沒食子酸甲酯、木犀草苷、蘆丁、鞣花酸、橄欖苦苷。
圖5為考察洗脫劑種類的基質固相分散萃取效果柱狀圖。圖中,1、2、3、4、5、6、7分別代表沒食子酸、羥基酪醇、沒食子酸甲酯、木犀草苷、蘆丁、鞣花酸、橄欖苦苷。
圖6為青果混合對照品的UHPLC-Q-TOF/MS色譜圖。圖中,1、2、3、4、5、6、7分別代表沒食子酸、羥基酪醇、沒食子酸甲酯、木犀草苷、蘆丁、鞣花酸、橄欖苦苷。
圖7為青果活性成分提取液的UHPLC-Q-TOF/MS色譜圖。
具體實施方式
下面結合附圖及具體實施例對本發明作進一步說明。
以下所述,僅是本發明的較佳實施例而已,但這些具體實施方式不以任何方式限制本發明的保護範圍。
實施例1
一、稱取50mg的青果粉末5份與25mg不同種類的殼聚糖(中分子殼聚糖、低分子殼聚糖、低粘度殼聚糖、高純度殼聚糖、羧化殼聚糖)分別加入五組研缽中,研磨60s。
二、將研磨好的固體作為填料裝入1ml規格的固相萃取小柱中,用0.5ml的甲醇對小柱洗脫三次,將收集到的洗脫液揮幹,再用200μl的洗脫液溶解,將離 心管內的液體渦旋10s後13000rpm離心5min。
三、取上層清液裝樣,用超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)採樣分析。
本實驗採用的儀器為安捷倫超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)。儀器:安捷倫超高效液相色譜系統(Agilent 1290 Infinity LC,Agilent Technologies,Santa Clara,CA,USA)配備真空抽氣泵,二元泵流動相系統,恆溫自動進樣器,恆溫柱溫箱。色譜柱Agilent Zorbax Extend-C18 column(2.1mm×50mm,1.8μm),柱溫40℃。流動相組成包括甲醇(A)和0.1%甲酸水溶液(B),梯度洗脫為0-1min:5%A,1-3.0min:5-20%A,3.0-4.0min:20-30%A,4.0-5.0min:30-30%A,5.0-6.0min:30-40%A,6.0-7.0min:40-45%A,7.0-8.0min:45-50%A,8.0-9.0min:50-80%A,9.0-10min:80-100%A,10-11min:100-100%A,11-12min:100-5%A,平衡時間5min。進樣體積1μL,流速0.4mL/min。
該UHPLC系統與下述質譜系統聯用:6530Q-TOF/MS(Agilent Technologies,Santa Clara,CA,USA)配備dual ESI離子源,儀器參數:負離子模式,霧化氣壓45psig,乾燥氣流速12L/min,乾燥氣溫度350℃,毛細管電壓3500V,裂解電壓175V,四極杆電壓65V,八極杆電壓750V,質譜檢測範圍為100-700m/z。數據解析軟體為Mass Hunter software(version B 05.00 Qualitative Analysis)。
不同種類吸附劑的萃取效果柱狀圖如圖2所示,結果表明,中分子殼聚糖的萃取效果最佳。可能由於它黏度、顆粒大小等適中能更好的與青果中的有效成分作用並能緊密地填在小柱內。故而,優選中分子殼聚糖作為吸附劑。
實施例2
一、稱取9份50mg的青果粉末於9組研缽中,並稱取8份不同質量的中分子殼聚糖(6.25mg,12.5mg,25mg,37.5mg,50mg,62.5mg,75mg,87.5mg)分別加入8組研缽中,剩下一組研缽不加吸附劑,研磨60s。
二、將研磨好的固體作為填料裝入1ml規格的固相萃取小柱中,用0.5ml的甲醇對小柱洗脫三次,將收集到的洗脫液揮幹,再用200μl的洗脫液溶解,將離心管內的液體渦旋10s後13000rpm離心5min。
三、取上層清液裝樣,用超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC- Q-TOF/MS)採樣分析。
不同青果與中分子殼聚糖的質量比的萃取效果如圖3中折線圖所示。由圖中可以看到隨著吸附劑量的增加萃取效果增強,基本增至25mg後萃取效果趨於平緩甚至略有下降,雖然25mg的中分子殼聚糖對物質1沒食子酸的萃取效果不是最佳,但綜合考慮,對總體來說是最優的。原因可能是,吸附劑用量的增加可以更多的吸附青果中的活性成分,但當活性成分基本被吸附之後,過多的吸附劑會使活性成分難以解吸,從而是萃取效果有所下降,
實施例3
一、稱取5份50mg的青果粉末和5份25mg的中分子殼聚糖放入5組研缽中,分別研磨不同的時間(20s,40s,60s,80s,100s)。
二、將研磨好的固體作為填料裝入1ml規格的固相萃取小柱中,用0.5ml的甲醇對小柱洗脫三次,將收集到的洗脫液揮幹,再用200μl的洗脫液溶解,將離心管內的液體渦旋10s後13000rpm離心5min。
三、取上層清液裝樣,用超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)採樣分析。
不同研磨時間的萃取效果折線圖如圖4所示。結果表明,隨著研磨時間的增加萃取效果增強,增至最優值60s後,隨著研磨時間的增加萃取效果反而有所下降。可能是由於充分的研磨有助於吸附劑更好的接觸青果中的活性成分,萃取效果增強,但是過度的研磨會使活性成分被擠壓到吸附劑中緻密的孔徑中,使得活性成分較難洗脫下來,從而使萃取效果下降。
實施例4
一、稱取6份50mg的青果粉末和6份25mg的中分子殼聚糖放入5組研缽中,研磨60s。
二、將研磨好的固體作為填料分別裝入6支1ml規格的固相萃取小柱中,分別用0.5ml不同的洗脫液(60%甲醇、80%甲醇、甲醇、乙腈、乙醇、乙酸乙酯)對小柱洗脫三次,將收集到的洗脫液揮幹,再用200μl的洗脫液溶解,將離心管內的液體渦旋10s後13000rpm離心5min。
三、取上層清液裝樣,用超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)採樣分析。
不同洗脫劑的萃取效果柱狀圖如圖5所示。結果顯示,隨著洗脫液極性的增強萃取效果逐漸加強,即60%的甲醇洗脫效果最佳,可能由於60%的甲醇極性與目標分子接近並能較好地溶解目標分子,且其洗脫時間相對較長,能與活性成分更好地接觸、相互作用,因此萃取效果最好。
日內精密度
一、稱取50mg的青果粉末和25mg的中分子殼聚糖放入研缽中,研磨60s。
二、將研磨好的固體作為填料裝入1ml規格的固相萃取小柱中,用0.5ml 60%的甲醇對小柱洗脫三次,將收集到的洗脫液揮幹,再用200μl的洗脫液溶解,將離心管內的液體渦旋10s後13000rpm離心5min。
三、取上層清液裝樣,用超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)採樣分析。在同一天內連續進樣6次。
日間精密度
一、稱取50mg的青果粉末和25mg的中分子殼聚糖放入研缽中,研磨60s。
二、將研磨好的固體作為填料裝入1ml規格的固相萃取小柱中,用0.5ml60%的甲醇對小柱洗脫三次,將收集到的洗脫液揮幹,再用200μl的洗脫液溶解,將離心管內的液體渦旋10s後13000rpm離心5min。
三、取上層清液裝樣,用超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)採樣分析。將該樣品連續進樣3天,每天2次。
日內、日間精密度實驗結果匯總如下表1:
表1
1,2,3,4,5,6,7分別代表沒食子酸、羥基酪醇、沒食子酸甲酯、木犀草苷、蘆丁、鞣花酸、橄欖苦苷。
重複性考察
一、稱取50mg的青果粉末和25mg的中分子殼聚糖放入研缽中,研磨60s。
二、將研磨好的固體作為填料裝入1ml規格的固相萃取小柱中,用0.5ml60%的甲醇對小柱洗脫三次,將收集到的洗脫液揮幹,再用200μl的洗脫液溶解,將離心管內的液體渦旋10s後13000rpm離心5min。
三、取上層清液裝樣,用超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)採樣分析。按上述步驟重複做三組,做為重複性考察。
藥材含量測定
一、稱取50mg的青果粉末和25mg的中分子殼聚糖放入研缽中,研磨60s。
二、將研磨好的固體作為填料裝入1ml規格的固相萃取小柱中,用0.5ml 60%的甲醇對小柱洗脫三次,將收集到的洗脫液揮幹,再用200μl的洗脫液溶解,將離心管內的液體渦旋10s後13000rpm離心5min。
三、取上層清液裝樣,用超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)採樣分析。獲得提取液的UHPLC-Q-TOF/MS色譜圖。
圖7為青果活性成分提取液的UHPLC-Q-TOF/MS色譜圖。
取沒食子酸、羥基酪醇、沒食子酸甲酯、木犀草苷、蘆丁、鞣花酸、橄欖苦苷的對照品適量,精密稱定,置於棕色量瓶中,加甲醇製成每1mL含沒食子酸250μg、羥基酪醇250μg、沒食子酸甲酯250μg、木犀草苷250μg、蘆丁250μg、鞣花酸250μg、橄欖苦苷250μg的對照品溶液。將製得的對照品溶液配置成一系列不同濃度的混合對照品溶液,按同樣的條件用UHPLC-Q-TOF/MS檢測,獲得7種對照品的色譜圖,以對照品的進樣量為橫坐標,對應色譜峰的峰面積為縱坐標,分別製作這7種物質的標準曲線。以青果提取液的色譜峰峰面積及標準曲線計算得到青果中沒食子酸、羥基酪醇、沒食子酸甲酯、木犀草苷、蘆丁、鞣花酸、橄欖苦苷的含量。
沒食子酸、羥基酪醇、沒食子酸甲酯、木犀草苷、蘆丁、鞣花酸、橄欖苦苷濃度均為100μg/mL的混合對照品溶液的色譜圖如圖6所示。
7種成分的標準曲線以及檢測限和定量限如下表2所示:
表2.
回收率實驗
一、稱取2份50mg的青果粉末和2份25mg的中分子殼聚糖分別放入2組研缽中,其中一組加入20μl100μg/ml的混和對照品溶液,研磨60s。
二、將研磨好的固體作為填料裝入1ml規格的固相萃取小柱中,用0.5ml 60%的甲醇對小柱洗脫三次,將收集到的洗脫液揮幹,再用200μl的洗脫液溶解,將離心管內的液體渦旋10s後13000rpm離心5min。
三、取上層清液裝樣,用超高效液相色譜-四極杆飛行時間質譜聯用儀(UHPLC-Q-TOF/MS)採樣分析。
重複性、含量測定、回收率實驗結果匯總如下表3:
表3
根據表中數據顯示,本發明方法的重複性好,回收率高,檢測準確性高。