一種靶向FAP‑alpha酶的抗腫瘤化合物及其製備方法與應用與流程
2023-12-10 06:11:27 3
本發明涉及一種化合物及其製備方法,尤其是一種靶向FAP-alpha酶的抗腫瘤化合物及其製備方法與應用,屬於抗腫瘤藥物領域。
背景技術:
當前,腫瘤的臨床化療藥物主要為細胞毒性藥物,儘管它們具備較強的抗腫瘤活性,然而由於這類化療藥物缺乏選擇性,往往導致嚴重的毒副作用。所以,實現細胞毒性藥物的靶向投遞對減少腫瘤化療的不良反應具有顯著的意義。
丙卡巴肼(procarbazine,Pcb)臨床上主要用於治療何杰金氏病,對於其他惡性腫瘤如惡性淋巴瘤、肺癌、多發性骨髓瘤等也有一定的療效。1982年該藥的鹽酸鹽以口服膠囊劑型被FDA批准在美國上市,主要用於惡性淋巴瘤的治療。然而由於這類化療藥物缺乏抗癌選擇性,往往導致較大的毒副作用,尤其對生殖細胞的毒化作用及骨髓抑制。眾所周知,骨髓抑制可導致白細胞、血小板減少,有出血傾向,亦可致貧血。這些毒副作用限制了其在臨床上的應用。
解決抗腫瘤藥物毒副作用的有效途徑之一是對藥物進行結構修飾,使之成為前體藥物,利用腫瘤細胞與正常細胞之間分子生物學上的差異,選擇性作用於腫瘤靶細胞的基因、酶、信號傳導因子等,從而達到靶向治療的目的。近年來大量的研究表明,成纖維細胞激活蛋白alpha(FAP-alpha)是由腫瘤相關成纖維細胞特異性高表達的腫瘤間質抗原分子,對腫瘤的發生發展有著重要的促進作用。FAP-alpha具有特異性肽鏈內切酶活性,能夠選擇性地水解N-末端封閉的甘氨醯脯氨酸二肽序列,如攜帶Z-Gly-Pro(Z-GP)二肽的底物。因此,選擇成纖維細胞激活蛋白alpha(FAP-alpha)作為靶標是提高腫瘤治療綜合指數的有效策略之一。
技術實現要素:
本發明的目的在於提供一種靶向FAP-alpha酶的抗腫瘤化合物,所述化合物能被FAP-alpha酶特異性水解切除二肽部分(Z-GP),釋放出丙卡巴肼,命名為Z-GP-丙卡巴肼(Z-GP-Pcb),所述Z-GP-Pcb能克服直接將丙卡巴肼作為抗癌藥物時具有嚴重毒副作用的不足,能顯著降低正常細胞的毒性和體內毒性,能顯著抑制荷瘤裸鼠體內腫瘤的生長以及降低對非靶器官毒性,有望開發成新的抗腫瘤藥物。
為實現上述目的,本發明採取的技術方案為:一種靶向FAP-alpha酶的抗腫瘤化合物,其結構如式I所示:
或者為式I所示化合物的同分異構體,其結構如式II所示:
所述化合物命名為Z-GP-丙卡巴肼(Z-GP-Pcb),式I和式II為同分異構體,兩者在藥物代謝、藥物用途中具有相同效果,均是腫瘤間質成纖細胞激活蛋白酶alpha(FAP-alpha)的特異性水解底物,其分子量為509.26Dα。
另外,本發明提供一種所述化合物Z-GP-丙卡巴肼的製備方法,所述方法包括以下步驟:
(1)將丙卡巴肼、苄氧羰基甘氨醯脯氨酸(Z-GP-OH)和縮合試劑,在0℃~30℃下攪拌並反應,得混合物;
(2)反應完畢後,向步驟(1)所得混合物中加入飽和鹽溶液,進行淬滅、分離、純化,即得化合物Z-GP-丙卡巴肼。
作為本發明所述化合物的製備方法的優選實施方式,所述步驟(1)中的縮合試劑為1-羥基苯並三唑和N,N-二異丙基乙胺和2-(7-氧化苯並三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯、氯甲酸乙酯、1-乙基-(3-二甲基氨基丙基)碳醯二亞胺鹽酸鹽、N,N』-二異丙基碳二亞胺、六氟磷酸苯並三唑-1-基-氧基三吡咯烷基磷、1-氯N,N,2-三甲基丙烯胺中的至少一種的混合物。
作為本發明所述化合物的製備方法的最優選實施方式,所述步驟(1)中的縮合試劑為2-(7-氧化苯並三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(HATU)、1-羥基苯並三唑(HOBT)和N,N-二異丙基乙胺(DIPEA)的混合物。
作為本發明所述化合物的製備方法的優選實施方式,所述步驟(1)中丙卡巴肼、苄氧羰基甘氨醯脯氨酸和縮合試劑的摩爾比為1:(1~3.0):(1.05~3.0)。
作為本發明所述化合物的製備方法的優選實施方式,所述步驟(2)中的鹽為NaCl、KCl、NH4Cl、(NH4)2SO4中的至少一種。
作為本發明所述化合物的製備方法的優選實施方式,所述步驟(1)中的丙卡巴肼的合成路線為:
具體採用如下方法製備而成:
(a)將對甲基苯甲酸溶於有機試劑,加入過量二氯亞碸,於60-80℃油浴加熱回流1-3h,旋蒸除去過量二氯亞碸,得對甲基苯甲醯氯,然後將對甲基苯甲醯氯重新溶於有機試劑中,緩慢加入異丙胺,控制溫度在30℃下,加料完畢繼續在室溫下攪拌反應2-4h,然後升溫至40℃,繼續反應20-40min,反應結束後分離純化,得N-異丙基對甲基苯甲醯胺;
(b)將步驟(a)所得的對甲基苯甲醯氯溶於有機試劑,緩慢加入硝酸鈰銨混懸液,加料完畢,置於90-100℃油浴鍋中反應20-30h,反應結束後分離純化,得4-甲醯基-N-異丙基苯甲醯胺;
(c)將步驟(b)所得的N-異丙基對甲基苯甲醯胺與甲基肼硫酸鹽溶於有機試劑,加入三乙胺,將反應裝置置於50-70℃油浴鍋中反應5-7h,旋幹溶劑,將混合物重新溶於有機試劑中,在0℃的低溫恆溫器中緩慢加入氰基硼氫化鈉,加料完畢,反應體系逐漸升至室溫,反應過夜,反應結束後分離純化,即得丙卡巴肼。
作為本發明所述化合物的製備方法的優選實施方式,所述Z-GP-丙卡巴肼的合成路線為:
作為本發明所述化合物的製備方法的優選實施方式,所述步驟(a)中的有機溶劑為乾燥的二氯甲烷或氯仿,所述對甲基苯甲酸與二氯亞碸的摩爾比為1.0:(5.0~100),所述對甲基苯甲酸與異丙胺的摩爾比為1:(5.0~10)。
作為本發明所述化合物的製備方法的優選實施方式,所述步驟(b)中的有機溶劑為硝酸溶液或醋酸溶液,所述硝酸鈰銨混懸液中硝酸溶液的濃度為3.0-5.0mmol/L,所述N-異丙基對甲基苯甲醯胺與硝酸鈰的摩爾投料比為1:(3.0~5.0)。
作為本發明所述化合物的製備方法的優選實施方式,所述步驟(c)中的有機溶劑為無水乙醇或DMF,所述4-甲醯基-N-異丙基苯甲醯胺與甲基肼硫酸鹽的摩爾比為1:(3.0~5.0)。
另外,本發明提供一種靶向FAP-alpha酶的抗腫瘤化合物,所述化合物為上述所述化合物Z-GP-丙卡巴肼的衍生物或生理上可接受的鹽。Z-GP-丙卡巴肼或Z-GP-丙卡巴肼生理上可接受的鹽在藥用中可以游離態形式存在。
作為本發明上述所述化合物的優選實施方式,所述化合物Z-GP-丙卡巴肼生理上可接受的鹽為酒石酸鹽、硫酸鹽、鹽酸鹽。優選地,所述Z-GP-丙卡巴肼生理上可接受的鹽的製備方法包括以下步驟:將Z-GP-丙卡巴肼溶於含1.05~3.0摩爾量酸(HA)的有機溶劑中,於-10℃~40℃溫度下攪拌反應3~20小時,分離固體化合物,洗滌,再將固體化合物重新溶於水中,冷凍乾燥,即得化合物Z-GP-丙卡巴肼生理上可接受的鹽。優選地,上述所述有機溶劑為體積比為1:1的甲醇和二氯甲烷的混合溶液。
另外,本發明提供一種藥物組合物,所述藥物組合物包括上述所述化合物Z-GP-丙卡巴肼和/或Z-GP-丙卡巴肼的衍生物和/或或Z-GP-丙卡巴肼在生理上可接受的鹽。
另外,本發明提供一種上述所述化合物或所述藥物組合物在製備抗腫瘤藥物中的應用。
作為本發明所述應用的優選實施方式,所述化合物或藥物組合物為腫瘤間質成纖細胞激活蛋白酶α的特異性水解底物。
作為本發明所述應用的優選實施方式,所述腫瘤為惡性淋巴瘤、肺癌、肝癌或多發性骨髓瘤。
本發明的有益效果在於:(1)本發明所述Z-GP-丙卡巴肼能顯著降低正常細胞的毒性和體內毒性,在體內外能被FAP-alpha酶特異性水解切除二肽部分(Z-GP)釋放出肼解產物;(2)所述Z-GP-丙卡巴肼能顯著抑制荷瘤裸鼠體內腫瘤的生長以及降低對非靶器官毒性;(3)本發明所述Z-GP-丙卡巴肼的製備方法具有反應條件溫和、實驗步驟簡單、收率高、產品純度高、經濟實用等特點。
附圖說明
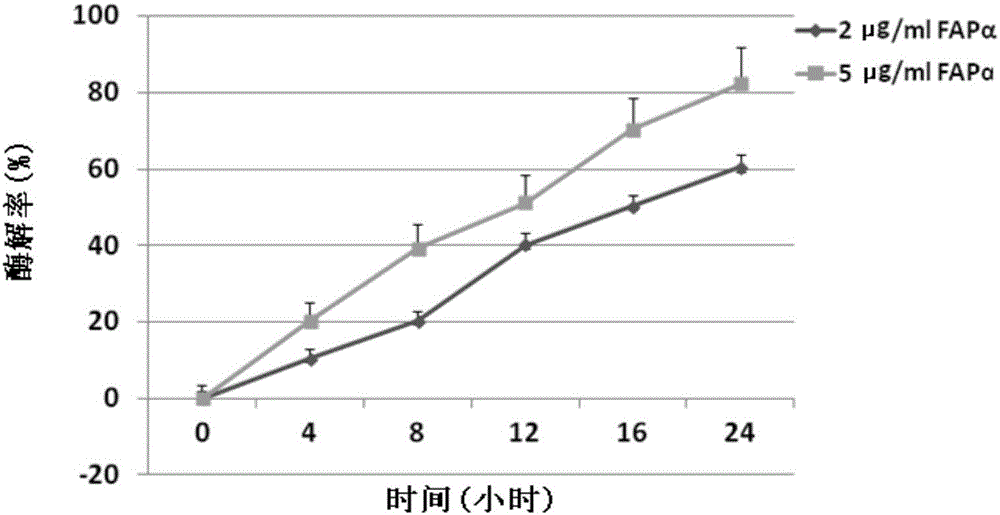
圖1是FAP-alpha對本發明所述化合物Z-GP-丙卡巴肼的酶解效應圖;
圖2是Pcb和Z-GP-Pcb對NCI-H460的細胞毒性作用影響對比圖,其中,與空白組相比,*P<0.05,**P<0.01;
圖3是分別採用FAP-alpha+Z-GP-Pcb共處理及Pcb處理對NCI-H460的細胞毒性作用影響對比圖,與空白組相比,*P<0.05,**P<0.01;
圖4是Pcb和Z-GP-Pcb對小鼠睪丸重量影響的對比圖,其中,與空白組相比,##P<0.01,與Pcb組相比,**P<0.01;
圖5是Pcb和Z-GP-Pcb對小鼠精子數量影響的對比圖,其中,與空白組相比,#P<0.05,與Pcb組相比,*P<0.05;
圖6是Pcb和Z-GP-Pcb在腫瘤及睪丸中的藥物分布比較圖,其中(A)為藥物在腫瘤組織的蓄積情況,(B)為藥物在睪丸組織的蓄積情況;
圖7是Pcb和Z-GP-Pcb對H22肝癌荷瘤小鼠腫瘤體積大小影響的對比圖。
具體實施方式
為更好的說明本發明的目的、技術方案和優點,下面將結合附圖及具體實施例對本發明作進一步說明。
實施例1
本發明所述Z-GP-丙卡巴肼的製備方法的一種實施例,所述製備方法包括以下步驟:
1.1.1 N-異丙基對甲基苯甲醯胺的製備
稱取對甲苯甲酸273mg(2.0mmol)於25mL的圓底燒瓶中,加入5.0mL(68.8mmol)二氯亞碸(過量),於78℃油浴加熱回流2h;旋蒸除去過量的二氯亞碸,得對甲基苯甲醯胺;將對甲基苯甲醯胺重新溶於2.0mL乾燥的二氯甲烷(DCM)中;另取1.0mL(11.7mmol)異丙胺溶於2.0mL乾燥的DCM中,得A液;控制溫度在30℃下,逐漸將A液滴加到反應體系中,加料完畢繼續攪拌30min,室溫下攪拌反應3h;之後升溫至40℃,繼續反應30min,停止加熱;將反應液傾入50mL冰水中,用二氯甲烷-乙醚(體積比1∶5)萃取4次,合併有機相,依次用水、稀鹽酸、水、稀氫氧化鈉溶液、水洗滌,無水硫酸鎂乾燥;旋蒸除去溶劑,得白色固體化合物289.8mg,收率88.9%。
經分析檢測,所得白色固體化合物的1H NMR(300MHz,CDCl3)δ:7.67(d,J=3.0Hz,1H),7.64(d,J=3.0Hz,1H),7.21(d,J=3.0Hz,1H),7.19(d,J=3.0Hz,1H),4.22~4.33(m,1H),2.38(s,3H),1.26(s,3H),1.24(s,3H);13C NMR(75MHz,CDCl3)δ:166.6,141.5,132.1,129.1,129.1,126.8,126.8,41.7,22.8,21.3,21.3.ESI-MS m/z:178.2[M+H]+,200.3[M+Na]+。以上數據證明所得白色固體產物為N-異丙基對甲基苯甲醯胺。
1.1.2 4-甲醯基-N-異丙基苯甲醯胺的製備
取5mL重蒸餾水與6mL濃硝酸混合均勻,製成約3.5mol/L的硝酸溶液備用;稱取N-異丙基對甲基苯甲醯胺177mg(1.0mmol)於耐壓管中,加入2mL 3.5mol/L硝酸溶液,超聲震蕩溶解;取硝酸鈰銨4.4g(4.0mmol)於西林瓶中,加入8.0mL 3.5mol/L硝酸溶液,攪拌5min,即得硝酸鈰銨的硝酸混懸液;取攪拌均勻的硝酸鈰銨混懸液逐滴滴加到耐壓管中,加料完畢,置於100℃油浴鍋中反應24h;反應結束,將反應液冷卻,隨後加入100mL飽和NaCl水溶液,用DCM萃取(3×100mL),合併有機相,加入無水硫酸鈉乾燥,過濾,旋幹有機溶劑得微黃色固體粉末,用矽膠柱色譜純化(乙酸乙酯-石油醚,體積比1:5),得白色固體136.2mg,收率71.2%。
經分析檢測,所得白色固體化合物的1H NMR(300MHz,CD3OD)δ:10.03(s,1H),7.95(d,J=9.0Hz,2H),7.58(d,J=9.0Hz,2H),4.10~4.30(m,1H),1.26(d,J=6.0Hz,3H),1.24(d,J=6.0Hz,3H);13C NMR(75MHz,CDCl3)δ:193.5,168.4,130.6,129.0,128.1(2),127.2(2),43.4,22.5(2);ESI-MS(m/z):192.3[M+H]+,224.5[M+Na]+。以上數據證明所得白色固體產物為4-甲醯基-N-異丙基苯甲醯胺。
1.1.3丙卡巴肼(procarbazine)的製備
稱取191mg(1.0mmol)4-甲醯基-N-異丙基苯甲醯胺和505mg(3.5mmol)甲基肼硫酸鹽於250mL反應瓶中,加入無水乙醇20.0mL,攪拌溶解,攪拌20min後,加入三乙胺1.0mL;將反應裝置置於60℃油浴鍋中反應6h,反應結束,旋幹溶劑,加入10.0mL的DMF將其重新溶解,在0℃的低溫恆溫器中緩慢加入氰基硼氫化鈉126mg(2.0mmol),加料完畢,反應體系逐漸升至室溫,反應過夜;反應結束後,加水淬滅反應;加入200.0mL的飽和NaCl水溶液,用200.0mL的乙酸乙酯萃取5次,合併有機相,加入無水硫酸鈉乾燥,過濾,合併有機相,旋幹得黃色油狀液體;矽膠柱色譜分離(乙酸乙酯-石油醚,體積比1:3),得丙卡巴肼161.9mg,收率74%。
經分析檢測,所得丙卡巴肼的1H NMR(300MHz,CDCl3)δ:7.47(d,J=7.0Hz,1H),7.43(d,J=7.0Hz,1H),7.31(d,J=7.0Hz,1H),7.24(d,J=7.0Hz,1H),3.88-3.91(m,1H),3.93(s,2H),2.47(s,3H),1.25(d,J=6.0Hz,3H),1.23(d,J=6.0Hz,3H);13C NMR(75MHz,CDCl3)δ:166.2,144.4,130.1,125.8,125.8,125.7,125.7,55.2,40.4,33.7,22.3,22.3;ESI-MS(m/z):222.3[M+H]+,244.3[M+Na]+。以上數據證明所得產物確實為丙卡巴肼(procarbazine)。
1.1.4 Z-GP-丙卡巴肼(Z-GP-Pcb)的製備
稱取612mg(2mmol)Z-GP-OH,283mg(2.1mmol)HOBt和798mg(2.1mmol)HATU溶於10mL乾燥DCM,將其倒入50mL圓底燒瓶中,加入二異丙基乙胺(DIPEA)0.3mL,將反應瓶置於冰浴中攪拌活化30min;另稱取442mg(2.0mmol)丙卡巴肼溶於5mL乾燥DCM,將其倒入西林瓶中,加入DIPEA 0.5mL,反應30min後,將溶液緩慢滴加到活化的反應瓶中,隨後將反應瓶移至室溫反應3-4h;反應完畢後加飽和NaCl溶液淬滅,以DCM萃取多次,合併有機相,依次用水、飽和NaCl溶液洗滌,無水Na2SO4乾燥後減壓蒸除溶劑;所得混合物經矽膠柱色譜(氯仿-甲醇,體積比10:1)和RP-HPLC分離純化(淋洗劑為MeOH:Water=50:50,V/V),得淡黃色黏稠狀液體690mg,收率65%。
經分析檢測,所得淡黃色黏稠狀液體的1H NMR(300MHz,CD3OD)δ:7.83~7.78(m,2H),7.52~7.47(m,2H),7.38~7.30(m,5H),5.10(d,J=6.6Hz,2H),4.29~3.96(m,5H),3.57(m,2H),3.22(s,1H),3.17(s,2H),2.05(m,3H),1.87~1.66(m,2H),1.26(d,J=6.7Hz,6H);13C NMR(75MHz,CD3OD)δ:173.58,166.38,165.55,156.31,140.79,137.39,134.46,129.11,128.77(2),128.31,127.75,127.71,127.48,127.27(2),65.88,65.84,56.59,52.32,48.91,45.92,41.43,29.76,24.29,21.89(2);ESI-MS(m/z):532.5[M+Na]+,以上數據證明所得淡黃色黏稠狀液體產物確實為Z-GP-丙卡巴肼(Z-GP-Pcb),其結構如式I所示:
或者為式I所示化合物的同分異構體,其結構如式II所示:
實施例2 Z-GP-Pcb靶向釋放特性的考察實驗
實驗方法:HPLC色譜條件如下:高效液相色譜儀Agilent 1200;色譜柱Cosmosil C18反相色譜柱(4.6×250mm,5μm);流動相(0min,55%甲醇和45%水(含2mM甲酸銨);10min,65%甲醇和35%水(含2mM甲酸銨);15min,75%甲醇和25%水(含2mM甲酸銨);30min,85%甲醇和15%水(含2mM甲酸銨);40min,85%甲醇和15%水(含2mM甲酸銨);流速1mL/min;檢測波長:254nm;進樣量2μL。
建立Z-GP-丙卡巴肼檢測標準曲線方法如下:將Z-GP-丙卡巴肼溶解於酶解緩衝液(50mM Tris-HCl,1.0M NaCl,pH 7.4),設置5個濃度梯度分別為0.012725、0.006363、0.003181、0.001591、0.000795μg/ml,以峰面積為縱坐標,藥物濃度為橫坐標繪製標準曲線,本實驗重複3次,將終濃度為30mM的Z-GP-丙卡巴肼孵育於含2μg/ml和5μg/ml的rhFAP-alpha的緩衝液中,反應溫度為37℃,在孵育0、4、8、12、16、24h時間點,取上清液用HPLC法檢測游離丙卡巴肼。
實驗結果:酶解實驗結果表明,FAP-alpha能催化Z-GP-Pcb水解釋放Pcb,且其酶解效率與酶濃度呈正相關(圖1)。
實施例3 Z-GP-Pcb酶解前後細胞毒性變化的考察實驗
實驗方法:將Z-GP-Pcb與Pcb分別在0.1-50μM的藥物濃度範圍內對NCI-H460細胞株(購於中國科學院上海細胞庫)處理48h,採用MTT法比較兩者的細胞毒性;將Z-GP-Pcb與FAP-alpha共孵育,取孵育上清液處理NCI-H460細胞株48h,採用MTT法分析FAP-alpha酶解作用對Z-GP-Pcb細胞毒性的影響,在FAP-alpha酶解作用下Z-GP-Pcb是否可恢復其潛在的細胞毒性。
實驗結果:細胞毒性實驗結果表明,Z-GP-Pcb對NCI-H460的細胞毒性明顯低於Pcb(圖2);在FAP-alpha酶解作用下,Z-GP-Pcb恢復其細胞毒作用(圖3)。
實施例4 Z-GP-Pcb精子毒性研究實驗
實驗方法:18-22g雄性昆明小鼠60隻,隨機分為5組,分別腹腔注射75mM Pcb/Z-GP-Pcb和150mM Pcb/Z-GP-Pcb,18天後取左側睪丸,稱重(g),加入生理鹽水製備精子懸液,吸取製備好的上清精子懸液移入另一離心管中,將其置於60~80℃的水浴中使精子死亡;取懸液1滴,滴入細胞計數板,計數懸液中的精子數,精子數=懸液精子數×稀釋倍數。
實驗結果:從表1和表2中可知,與對照組比較,75mM和150mM的丙卡巴肼分別單次注射18天後,睪丸重量分別降低29.5%和55.4%;精子數分別顯著降低42.2%和52.7%,差異有統計學意義;而Z-GP-Pcb 75mM和150mM分別單次注射18天後,與對照組相比,睪丸重量幾乎無變化,精子數量降低較不明顯,分別為33.4%和35.3%,Z-GP-Pcb分別與對應劑量丙卡巴肼比較,睪丸重量和精子數量明顯增加,有統計學差異(圖4和圖5)。
表1 Z-GP-Pcb對小鼠睪丸重量的影響
註:與空白組相比,##P<0.01,與Pcb組相比,**p<0.01
表2 Z-GP-Pcb對小鼠精子數量影響
註:與空白組相比,#P<0.05,與Pcb組相比,*P<0.05
實施例5 Z-GP-Pcb靶向分布特性考察實驗
實驗方法:取KM小鼠150隻,無菌吸取傳代H22瘤株(購自於中國醫學科學院腫瘤細胞庫)且生長良好的小鼠腹水,用生理鹽水稀釋成1*107cells/mL,每鼠0.2mL接種於小鼠右側腋窩皮下建立荷瘤小鼠模型;對模型小鼠(n=10)一次性靜脈給予10mg/kg Z-GP-Pcb或Pcb,分別在給藥後0.25,2,8h三個時間點處死小鼠,採集腫瘤和睪丸組織,通過HPLC法檢測其組織的藥物濃度。
實驗結果:藥物組織分布結果顯示,在給藥1h內Z-GP-Pcb在荷瘤小鼠睪丸的藥物蓄積顯著低於Pcb(P<0.01),且Z-GP-Pcb在給藥0.25,1,4h時,其腫瘤的蓄積比率均明顯高於Pcb(圖6)。
實施例6 Pcb和Z-GP-Pcb抗小鼠H22肝癌作用實驗
實驗方法:取KM小鼠60隻,無菌吸取傳代H22瘤株且生長良好的小鼠腹水,用生理鹽水稀釋成1*107cells/mL,每鼠0.2mL接種於小鼠右側腋窩皮下,建立荷瘤小鼠模型;接種24h後,將成功接種的KM小鼠,隨機分為以下6組,每組10隻,造模後第二天開始給藥,受試藥物組每天給藥,給藥方式為腹腔注射給藥10mL/kg,連續給藥14天,末次給藥24h後處死小鼠並檢測相關指標。
測試指標:隔日稱量動物體重;末次給藥後眼球取血,血球儀計算白細胞、血紅蛋白、血小板等;瘤重及抑瘤率:取動物瘤組織稱瘤重;
按下式計算抑瘤率:
抑瘤率=(模型組瘤重-給藥組瘤重)/模型組瘤重×100%
臟器指數:取肝臟、胸腺及脾臟,按下式計算肝臟、胸腺及脾重指數:
臟器指數=臟器重量(mg)/體重(g)
實驗結果:從圖7、表3和表4中可知,Pcb高劑量、Pcb低劑量、Z-GP-Pcb高劑量、Z-GP-Pcb低劑量組抑瘤率分別為:73.91%、33.93%、69.81%、35.39%,從受試藥物抑瘤率看,這2個受試藥物都表現出比較好的抗腫瘤作用,等摩爾劑量的Pcb和Z-GP-Pcb抑瘤效果相當,另外除Pcb高劑量組動物體重下降明顯外,其他各組體重沒有顯著變化;Pcb和Z-GP-Pcb兩個樣品對脾臟指數和胸腺指數影響不大;Pcb高、低劑量也能夠使動物白細胞計數、血小板含量減少,明顯產生骨髓抑制作用;而Z-GP-Pcb高、低劑量對血細胞數量無明顯影響;說明Z-GP-Pcb對非靶器官毒性明顯降低,改善了Pcb致白細胞和血小板減少的不良反應。
表3 Pcb、Z-GP-Pcb對H22肝癌荷瘤小鼠的影響
註:與模型組比較(降低),*P<0.05,**P<0.01
表4 Pcb和Z-GP-Pcb對H22肝癌荷瘤小鼠血細胞的影響
註:與模型組比較(降低),*P<0.05;**P<0.01;Z-GP-Pcb低劑量組與相應Pcb低劑量組比較(升高),#P<0.05;Z-GP-Pcb高劑量組與相應Pcb高劑量組比較(升高),##P<0.01
以上結果揭示了本發明所述Z-GP-Pcb能顯著降低正常細胞的毒性和體內毒性,在體內外能被FAP-alpha酶特異性水解切除二肽部分(Z-GP),釋放出丙卡巴肼,本發明所述化合物能顯著抑制荷瘤裸鼠體內腫瘤的生長以及降低對非靶器官毒性,有望開發成新藥。
最後所應當說明的是,以上實施例僅用以說明本發明的技術方案而非對本發明保護範圍的限制,儘管參照較佳實施例對本發明作了詳細說明,本領域的普通技術人員應當理解,可以對本發明的技術方案進行修改或者等同替換,而不脫離本發明技術方案的實質和範圍。