一種水相「一鍋法」合成含異噁唑環結構的物質的方法與流程
2023-05-16 08:34:46 2
本發明涉及有機合成技術領域,具體為一種水相「一鍋法」合成含異噁唑環結構的物質的方法。
背景技術:
雜環化合物是提供藥物核心骨架的重要物質,異噁唑是一種含氮雜環結構的化合物,廣泛用於在有機合成中,是一種重要的有機合成中間體。另外,異噁唑環也是許多抗糖尿病活性分子的重要骨架結構,其衍生物具有葡萄糖苷酶抑制活性,例如:化合物(A),(B),(C)已顯示出了一定的抗糖尿病活性。此外,一些含異噁唑環結構的物質具有抑制雜草和土壤細菌生長的效能,因此它在農藥和殺蟲劑領域具有廣泛的應用。
綠色化學是近現代才發展起來的一門重要化學分支,其已在整個化學領域備受關注,是當代解決環境汙染問題和實現可持續發展的有效途徑,是對環境友好型的化學研究方向。綠色化學的根本目是減少資源消耗和避免有危害的化合物的產生,達到高效,零汙染,可持續,經濟的目標。據所查文獻,之前關於合成含異噁唑環結構物質的相關報導,其合成大都在有機溶劑中進行,且利用無機酸作為催化劑,這些會對環境造成汙染,無法達到可持續發展的目的。綠色化學合成要求反應過程中採用無毒無害的試劑、溶劑或催化劑,以達到經濟、可持續的應用的要求。而其中水又被認為是最理想的溶劑,因此,以綠色化學的概念來看,開發具有價格低廉、無毒無害和環境友好的方法合成含異噁唑環結構的物質具有重要的意義。
技術實現要素:
本發明的目的在於提供一種水相「一鍋法」合成含異噁唑環結構的物質的方法,它能有效的解決背景技術中存在的問題。
為實現上述目的,本發明提供如下技術方案:一種水相「一鍋法」合成含異噁唑環結構的物質的方法,包括結構式III所示的芳香醛、結構式IV所示的2-萘酚、結構式V所示的3-氨基-5-甲基異噁唑和結構式VI所示的苯乙酮;將結構式III所示的芳香醛與結構式V所示的3-氨基-5-甲基異噁唑,以及結構式IV所示的2-萘酚(或結構式VI所示的苯乙酮)為原料,在無機鹽存在下,利用水為溶劑,於70-95℃反應1-4h,冷卻至室溫後過濾,粗產品經重結晶即得到產物,其反應通式如下:
其中R1為苯基或取代苯基;分離純化方式可採用過濾後重結晶等。
進一步,所述R1為苯基,對甲基苯基、對甲氧基苯基、對氯苯基、間硝基苯基。
進一步,所述結構式III所示的芳香醛與結構式IV所示的2-萘酚(或結構式VI所示的苯乙酮)的摩爾比為1:1—1:2,優選為1:1;所述結構式III所示的芳香醛與所述結構式V所示的3-氨基-5-甲基異噁唑的摩爾比為1:1。
進一步,所述無機鹽為氯化鈉、硝酸鈉、氯化鋰、硝酸鉀或硫酸鈉中的一種,優選為氯化鋰。
進一步,每1mmol結構式III所示的芳香醛,所需的溶劑水為3mL。
進一步,結構式III所示的芳香醛與無機鹽的摩爾比為1:5—1:8,優選為1:6。
進一步,所述反應溫度在70-95℃間,優選反應溫度為90℃。
進一步,所述反應時間為3h。
與現有技術相比,本發明的有益效果是:本發明方法避免了用無機酸做催化劑時腐蝕設備、汙染環境的不足之處,反應溶劑環境友好,易於得到,且終產物容易分離純化、反應收率較高,達到80%以上。
附圖說明
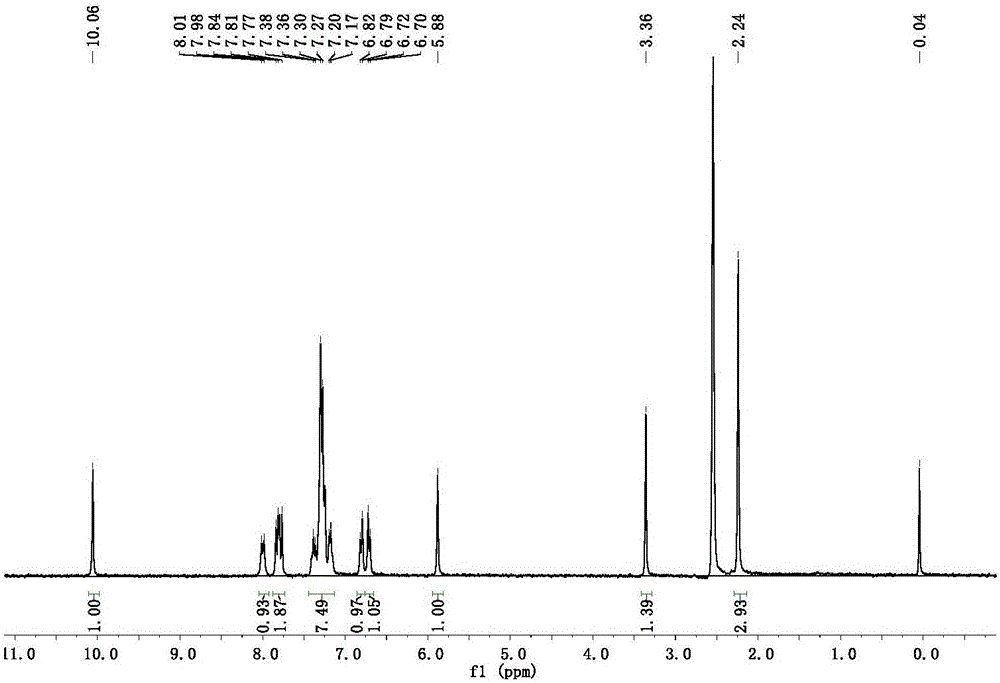
圖1為實施例1中化合物I-a的1H NMR圖;
圖2為實施例1中化合物I-a的13C NMR圖。
具體實施方式
下面將結合本發明實施例中的附圖,對本發明實施例中的技術方案進行清楚、完整地描述,顯然,所描述的實施例僅僅是本發明一部分實施例,而不是全部的實施例。基於本發明中的實施例,本領域普通技術人員在沒有做出創造性勞動前提下所獲得的所有其它實施例,都屬於本發明保護的範圍。
一種水相「一鍋法」合成含異噁唑環結構的物質的方法,包括結構式III所示的芳香醛、結構式IV所示的2-萘酚、結構式V所示的3-氨基-5-甲基異噁唑和結構式VI所示的苯乙酮;將結構式III所示的芳香醛與結構式V所示的3-氨基-5-甲基異噁唑,以及結構式IV所示的2-萘酚(或結構式VI所示的苯乙酮)為原料,在無機鹽存在下,利用水為溶劑,於70-95℃反應1-4h,冷卻至室溫後過濾,粗產品經重結晶即得到產物,其反應通式如下:
其中R1為苯基或取代苯基;分離純化方式可採用過濾後重結晶等;所述R1為苯基,對甲基苯基、對甲氧基苯基、對氯苯基、間硝基苯基;所述結構式III所示的芳香醛與結構式IV所示的2-萘酚(或結構式VI所示的苯乙酮)的摩爾比為1:1—1:2,優選為1:1;所述結構式III所示的芳香醛與所述結構式V所示的3-氨基-5-甲基異噁唑的摩爾比為1:1;所述無機鹽為氯化鈉、硝酸鈉、氯化鋰、硝酸鉀或硫酸鈉中的一種,優選為氯化鋰;每1mmol結構式III所示的芳香醛,所需的溶劑水為3mL;結構式III所示的芳香醛與無機鹽的摩爾比為1:5—1:8,優選為1:6;所述反應溫度在70-95℃間,優選反應溫度為90℃;所述反應時間為3h。
實施例一:
取0.14g(1mmol)2-萘酚,0.1mL(1mmol)苯甲醛和0.10g(1mmol)3-氨基-5-甲基異噁唑,並將它們加入到裝有攪拌子的10mL反應瓶中,再向其中加入0.50g(6mmol)LiCl,3mL蒸餾水。將混合物升溫至90℃,並在此溫度反應3h,瓶中有淡紅色固體析出。待此混合物靜置冷卻至室溫,過濾,用水洗滌,得粗產品,用丙酮重結晶,乾燥,得白色顆粒晶體,產率90%。
1H NMR(400MHz,DMSO-d6,TMS)δ:10.06(s,1H),8.0(d,J=9.0Hz,1H),7.84-7.77(m,2H),7.38-7.17(m,7H),6.82-6.70(m,2H),5.88(s,1H,-CHNH),3.3(s,1H,-NH),2.24(s,3H,-CH3);13C NMR(100MHz,DMSO-d6,TMS)δ:168.00,165.19.153.41,143.86,132.85,129.79,129.26,129.20,128.59,126.86,126.69,124.38,123.05,122.84,120.64,119.08,94.68,53.26,12.74。
得到的白色顆粒晶體的結構式為:(I-a)
實施例二:
取2.09g(5mmol)對甲基苯甲醛,0.73g(5mmol)2-萘酚和0.50g(5mmol)3-氨基-5-甲基異噁唑,並將它們加入到裝有攪拌子的50mL反應瓶中,再加入2.5g(30mmol)LiCl和15mL蒸餾水。將混合物升溫至90℃,並在此溫度反應3h。待此混合物靜置冷卻至室溫,過濾,用丙酮重結晶,得粉紅色顆粒晶體,產率82%。
1H NMR(400MHz,DMSO-d6)δ10.05(s,1H),8.02(d,J=7.7Hz,1H),7.81-7.75(m,2H),7.37(s,1H),7.30–7.16(m,4H),7.07(d,J=8.0Hz,2H),6.79(d,J=7.8Hz,1H),6.70(d,J=7.7Hz,1H),5.88(s,1H),2.24(s,3H),2.21(s,3H);13C NMR(100MHz,DMSO)δ167.89,165.21,153.02,140.64,135.49,132.80,129.52,129.09,128.96,127.85,126.63,126.59,124.06,122.77,120.59,118.93,94.39,52.92,21.66,13.27。
得到的粉紅色顆粒晶體的結構式為:(I-b)
實施列三:
取2.18g(5mmol)4-甲氧基苯甲醛,0.72g(5mmol)2-萘酚和0.50g(5mmol)3-氨基-5-甲基異噁唑,並將它們加入到50mL反應瓶中,再加入2.5g(30mmol)LiCl和15mL蒸餾水。將混合物升溫至90℃,並在此溫度反應3h。待此混合物靜置冷卻至室溫,過濾,用丙酮重結晶,得淡黃色顆粒晶體,產率75%。
1H NMR(400MHz,DMSO)δ10.05(s,1H),8.02(d,J=7.7Hz,1H),7.81-7.75(m,2H),7.37(s,1H),7.30–7.16(m,4H),7.07(d,J=8.0Hz,2H),6.79(d,J=7.8Hz,1H),6.70(d,J=7.7Hz,1H),5.88(s,1H),2.19(s,3H),2.08(s,3H);13C NMR(100MHz,DMSO)δ167.73,166.01,157.77,153.50,135.93,132.56,129.49,129.09,128.96,127.79,126.55,122.81,120.46,118.93,113.80,94.49,55.63,52.93,31.04,12.57。
得到的淡黃色顆粒晶體的結構式為:(I-c)
儘管已經示出和描述了本發明的實施例,對於本領域的普通技術人員而言,可以理解在不脫離本發明的原理和精神的情況下可以對這些實施例進行多種變化、修改、替換和變型,本發明的範圍由所附權利要求及其等同物限定。