一種基於氫鍵作用熱驅動自修復彈性體的製備方法與流程
2023-05-06 10:38:31 2
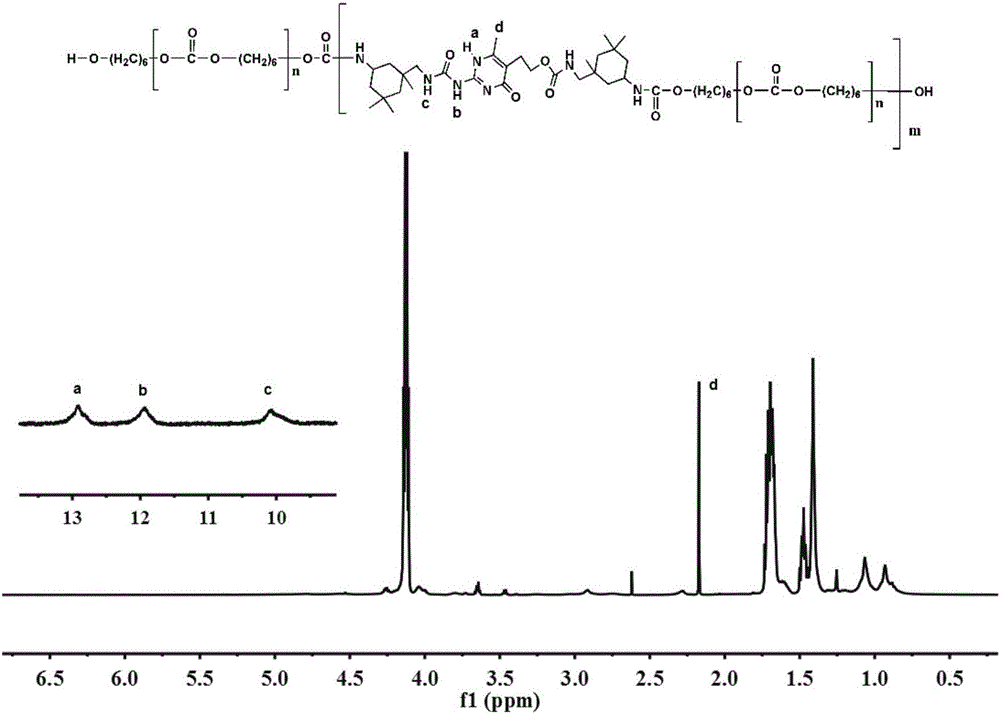
本發明涉及具有自修復功能的氫鍵本徵型聚氨酯彈性體材料的製備方法,具體涉及一種基於氫鍵作用熱驅動自修復彈性體的製備方法。
技術背景
當材料在受到外部機械作用時,容易對材料表面或內部造成損傷,使材料產生裂紋,對材料的力學性能及使用壽命造成嚴重影響。為解決材料在使用過程中容易產生損傷的問題,人們引入了自修復的概念。自修復材料作為一種新型的智能材料,在一些重要工程和特殊領域具有重要的發展前景及運用價值。
目前,研究較多的是使用包覆有修復劑的微膠囊自修復材料,這種修復材料雖具備一定的自修復能力,但是也存在缺點,如:每種修復劑對應的基體有限並且在材料的每處均只有一次修復能力;微膠囊在基材中的穩定性較差,不能長時間穩定存在於基體中;材料產生裂紋時不能每次保證微膠囊破裂釋放修復劑。基於氫鍵作用的自修復材料,因為氫鍵可逆的緣故使得材料在每一處均具有多次修復能力且材料易於加工;但是普通氫鍵的鍵能較低,製備的氫鍵聚合物材料性能較差。本發明為解決這一問題引入了一種具有自識別形成可逆四重氫鍵的脲基嘧啶酮單元(Upy),將該單元以封端的形式引入到聚氨酯低聚物的兩端製備了Upy封端聚氨酯材料以及以加聚反應的方式使Upy單元分布在聚合物鏈段中製備Upy單元個數可控的聚氨酯彈性體。對於Upy四重氫鍵作用自修復材料大部分是水凝膠方面的研究。目前已有研究採用Upy直接對聚酯二元醇、羥基矽油以及聚醚二元醇等封端製備的自修復材料存在強度低、彈性差、修復溫度高等缺點。雖然採用Upy對鏈段相對硬的PCDL進行封端製備的超分子聚合物能獲得較高的強度,但其伸長率低,彈性差,修復效率低且修復溫度高,如表1對比樣。為解決這些問題,本發明合成了羥基封端聚氨酯低聚物,再對其進行Upy封端處理,在保證材料強度的同時提高了材料的伸長率且有效降低了修復溫度。本發明還通過Upy型二異氰酸酯的合成使其與低聚物二元醇以加聚的形式分布在聚氨酯鏈中,且Upy單元數量可控,製備了高強度以及高伸長率的自修復聚氨酯彈性體,在較低溫度下即可達到出色的修復性能。
技術實現要素:
本發明的目的是提供一種基於可逆氫鍵作用自修復聚氨酯彈性體的製備方法,該方法通過將具有自識別形成可逆四重氫鍵的UPY單元引入到聚氨酯分子鏈兩端製備兩末端為Upy單元的遙爪聚合物(PU-Upy)或者引入到聚氨酯分子鏈端中間製備可形成氫鍵網絡結構的熱塑性聚合物(HPU-Upy)。該方法製備的熱塑性彈性體具有強度高、伸長率大並且能在相對低的溫度下達到90%以上的修復效率。
本發明的目的至少通過如下技術方案之一實現。
一種基於氫鍵作用熱驅動自修復彈性體的製備方法,包括如下步驟:
(1)將嘧啶酮類化合物與二異氰酸酯(摩爾比為1:7~1:12)在90~110℃及惰性氣體保護下攪拌反應18~24h後冷卻至室溫,加入正己烷或正庚烷得到白色沉澱,過濾並洗滌沉澱物,30~50℃真空乾燥24~40h得到脲基嘧啶酮型異氰酸酯;所述嘧啶酮類化合物為2-氨基-4-羰基-6-甲基嘧啶(MIS)或2-氨基-4-羰基-5-(2-羥乙基)-6-甲基嘧啶(HMIS);當所述嘧啶酮類化合物為2-氨基-4-羰基-6-甲基嘧啶(MIS)時,所述脲基嘧啶酮型異氰酸酯為脲基嘧啶酮型單異氰酸酯(Upy-NCO);當所述嘧啶酮類化合物為2-氨基-4-羰基-5-(2-羥乙基)-6-甲基嘧啶(HMIS)時,所述脲基嘧啶酮型異氰酸酯為脲基嘧啶酮型二異氰酸酯(NCO-Upy-NCO);
(2)當步驟(1)中,嘧啶酮類化合物為2-氨基-4-羰基-6-甲基嘧啶(MIS)時,其步驟如下:取低聚物二元醇與計量的二異氰酸酯在催化劑下及惰性氣體保護下40~50℃反應4~6h,製備端羥基聚氨酯低聚物,取計量的步驟(1)得到的脲基嘧啶酮型單異氰酸酯(Upy-NCO)與所述端羥基聚氨酯低聚物混合,並用無水溶劑溶解均勻,在惰性氣體保護下50-70℃反應18~24h,得到基於氫鍵作用熱驅動自修復彈性體;
當步驟(1)中,嘧啶酮類化合物為2-氨基-4-羰基-5-(2-羥乙基)-6-甲基嘧啶(HMIS)時,其步驟如下:取低聚物二元醇與計量的步驟(1)中得到的脲基嘧啶酮型二異氰酸酯(NCO-Upy-NCO)在催化劑以及惰性氣體保護下,40~70℃反應16-24h,得到基於氫鍵作用熱驅動自修復彈性體。
上述方法中,步驟(1)中所述二異氰酸酯包括六亞甲基二異氰酸酯(HDI)、異佛爾酮二異氰酸酯(IPDI)或二苯基甲烷二異氰酸酯(MDI)。
上述方法中,步驟(2)中所述的低聚物二元醇包括聚碳酸酯二醇(PCDL2000)、聚四亞甲基醚二醇(PTMG2000)、聚己內酯二醇(PCL2000)或聚乙二醇(PEG2000)中的一種以上。
上述方法中,步驟(2)中,所述二異氰酸酯包括六亞甲基二異氰酸酯(HDI)或異佛爾酮二異氰酸酯(IPDI)。
上述方法中,步驟(2)中,所述無水溶劑為無水甲苯、無水氯仿或無水乙酸乙酯。
上述方法中,步驟(2)中,端羥基聚氨酯低聚物的製備中低聚物二元醇與二異氰酸酯摩爾比控制為1:0~1:0.9。
上述方法中,步驟(2)中,所述低聚物二元醇與脲基嘧啶酮型二異氰酸酯的摩爾比值控制在1.6-1.1範圍,通過控制兩者的比值可調節鏈段中Upy單元的數量。
上述方法中,步驟(1)和步驟(2)中的反應均在惰性氛圍中進行,所述惰性氛圍為氮氣。
上述方法中,步驟(2)中,所述催化劑均為二月桂酸二丁基錫。
上述方法中,所述2-氨基-4-羰基-6-甲基嘧啶(MIS)與2-氨基-4-羰基-5-(2-羥乙基)-6-甲基嘧啶(HMIS)的結構如下:
所述脲基嘧啶酮型單異氰酸酯(Upy-NCO)與脲基嘧啶酮型二異氰酸酯(NCO-Upy-NCO)的結構如下:
1.PU-Upy合成步驟:
(1)脲基嘧啶酮型單異氰酸酯分子的合成:稱取10g 2-氨基-4-羰基-6-甲基嘧啶(MIS)於裝有回流冷凝管的三口燒瓶中,繼續加入80-100ml的二異氰酸於燒瓶中並在氮氣保護下升溫至100℃反應24h。反應結束後體系冷卻至室溫,加入200ml正己烷,過濾得到白色沉澱。採用正己烷重複洗滌並過濾除去殘餘的二異氰酸酯。所得白色沉澱於40℃下真空乾燥24h,得到白色粉末狀產物(Upy-NCO)。
(2)低聚物二元醇於120℃下真空乾燥4h待用,取40g乾燥過的低聚物二元醇於裝有回流裝置的燒瓶中,氮氣保護下升溫至50℃,加入計量的二異氰酸酯,直至紅外光譜中-NCO的特徵峰消失後加入100ml無水甲苯以及計量的Upy-NCO(殘餘羥基摩爾數的1.5倍)並升溫至100℃反應16小時左右,直至核磁氫譜中檢測不到羥基質子時停止反應。採用甲醇沉澱並洗滌得到白色沉澱物,真空乾燥後得到透明的彈性材料(PU-Upy)。
2.HPU-Upy的合成步驟:
(1)脲基嘧啶酮型二異氰酸酯分子的合成:稱取2-氨基-4-羰基-5-(2-羥乙基)-6-甲基嘧啶(HMIS)10g於裝有回流冷凝裝置的三口瓶中,加入80-100ml的二異氰酸酯,在氮氣保護下升溫至90℃,在磁力攪拌下反應40小時。反應結束後體系冷卻至室溫,加入200ml正庚烷沉澱產物,過濾並重複洗滌沉澱物,將沉澱物在40℃下真空乾燥24h得到白色粉末產物(NCO-Upy-NCO)。
(2)取40g經過真空乾燥過的低聚物二元醇於三口燒瓶中,升溫至60℃,在機械攪拌下緩慢滴加經100ml無水甲苯稀釋過NCO-Upy-NCO,整個反應過程在氮氣保護下進行。反應至紅外光譜檢測不到-NCO基團的吸收峰時結束反應。將溶液在常溫下揮發乾燥,得到透明彈性材料(HPU-Upy)。
與現有技術相比,本發明的優勢在於:
相比於現階段已存在的基於氫鍵作用自修復材料,本發明的氫鍵型自修復聚氨酯彈性體具備較高的拉伸強度以及斷裂伸長率,並且能夠在相對低的修復溫度下獲得出色的自修復性能。
附圖說明
圖1是PU2-Upy的核磁氫譜圖;
圖2為HPU1.2-Upy的核磁氫譜;
圖3是PCDL-Upy的核磁氫譜圖。
具體實施方式
下面結合具體實施例對本發明作進一步地具體詳細描述,但本發明的實施方式不限於此,對於未特別註明的工藝參數,可參照常規技術進行。
脲基嘧啶酮型單異氰酸酯分子(Upy-NCO)的製備方法如下:取2-氨基-4-羰基-6-甲基嘧啶(MIS)10g於三口燒瓶中,加入六亞甲基二異氰酸酯(HDI)100ml,在氮氣保護下升溫至100℃,磁力攪拌反應18h後停止反應並冷卻至室溫。加入200ml正己烷得到白色沉澱,對沉澱進行多次洗滌以除去殘餘的二異氰酸酯,過濾並在40℃下真空乾燥24h得到白色粉末產物(Upy-NCO)。
脲基嘧啶酮型二異氰酸酯(NCO-Upy-NCO)的製備方法如下:取2-氨基-4-羰基-5-(2-羥乙基)-6-甲基嘧啶(HMIS)10g於三口燒瓶中,加入100ml異佛爾酮二異氰酸酯(IPDI),在氮氣保護下升溫至90℃,磁力攪拌反應40h後結束反應並冷卻至室溫。加入200ml正庚烷,收集沉澱,再利用正庚烷多次洗滌沉澱得到白色固體,置於40℃的真空乾燥箱中乾燥24h後得到白色粉末產物(NCO-Upy-NCO)。
實施例1
取40g經過真空乾燥的分子量為2000的聚碳酸酯二醇(PCDL)及1滴催化劑(二月桂酸二丁基錫)於裝有回流冷凝管的三口燒瓶中,氮氣保護下升溫至50℃並緩慢滴加經10ml無水甲苯稀釋過的計量的六亞甲基二異氰酸酯,機械攪拌下直至紅外光譜中-NCO吸收峰消失後再加入150ml無水甲苯並升溫至100℃;加入Upy-NCO(體系殘餘羥基摩爾數的1.5倍),機械攪拌下反應16h左右取樣在核磁氫譜檢測下羥基質子峰消失時停止反應。冷卻至室溫後將聚合物溶液滴加到200ml正己烷中得到白色沉澱物,對沉澱多次洗滌乾燥後得到透明彈性固體。調節聚碳酸酯二醇與六亞甲基二異氰酸酯的摩爾比值分別為2和1.4,最終製備的彈性體分別為PU2-Upy及PU1.4-Upy。
對上述製備的彈性體進行表徵,圖1是PU2-Upy的核磁氫譜圖,修復效率見表1,其測試結果表明,PU2-Upy在70℃下修復4h,修復效率可高達90%;PU1.4-Upy在40℃下修復4h即可達到92%的修復效率。
實施例2
取40g經過真空乾燥的分子量為2000的聚碳酸酯二醇(PCDL)及1滴催化劑(二月桂酸二丁基錫)於裝有回流冷凝管的三口燒瓶中,氮氣保護下升溫至60℃並緩慢滴加經100ml無水甲苯溶解好的NCO-Upy-NCO,機械攪拌下直至紅外光譜中-NCO吸收峰消失後停止反應。在聚四氟乙烯槽中常溫揮發得到透明的彈性薄膜。調節PCDL與NCO-Upy-NCO的摩爾比值分別為1.2,得到聚氨酯彈性體HPU1.2-Upy。
對上述製備的彈性體進行表徵,圖2為HPU1.2-Upy的核磁氫譜,修復效率見表1,結果表明,HPU1.2-Upy在80℃下修復2h修復效率為71%,修復4h後修復效率高達91%,修復6h甚至能達到95%的修復效率。
對比例
採用Upy直接對PCDL封端雖然能獲得相對好的拉伸強度,但是材料存在彈性差,修復效率低等缺點;為突出本發明的優勢,製備了PCDL-Upy並對其修復性能進行測試,與本發明進行對比。
取40g經過真空乾燥的分子量為2000的聚碳酸酯二醇(PCDL)於裝有回流冷凝管的三口燒瓶中,加入100ml無水甲苯,在氮氣保護下升溫至100℃;加入17.58g Upy-NCO,機械攪拌下反應16h左右取樣在核磁氫譜檢測下羥基質子峰消失時停止反應。冷卻至室溫後將聚合物溶液滴加到200ml正己烷中得到白色沉澱物,對沉澱多次洗滌乾燥後得到透明彈性固體(PCDL-Upy)。
對上述製備的彈性體進行表徵,圖3是PCDL-Upy的核磁氫譜圖,薄膜修復效率見表1,其測試結果表明,PCDL-Upy薄膜在80℃下修復4h僅能達到75%的修復效率。
相關測試方法如下:
核磁氫譜制樣及測試方法:取10-20mg彈性體樣品溶解於含TMS的氘代氯仿溶劑中,採用600MHZ的核磁共振儀進行氫譜掃描。
拉伸性能測試參照國標QB/T2415-1998進行測試。
修復性能測試製樣及評估方法:採用剃刀在啞鈴狀標準樣條的中央處裁斷,將兩端面緊靠一起,置於恆溫箱中進行修復;樣條的修復效率定義為修復樣條與原試樣條的最大拉伸強度的比值。
表1樣品機械性能以及修復性能數據
本發明的上述實施例僅僅是為清楚地說明本發明所作的舉例,而並非是對本發明的實施方式的限定。對於所屬領域的普通技術人員來說,在上述說明的基礎上還可以做出其它不同形式的變化或變動。這裡無需也無法對所有的實施方式予以窮舉。凡在本發明的精神和原則之內所作的任何修改、等同替換和改進等,均應包含在本發明權利要求的保護範圍之內。