一種鹽酸蘭地洛爾的製備方法與流程
2023-06-02 12:42:36 5
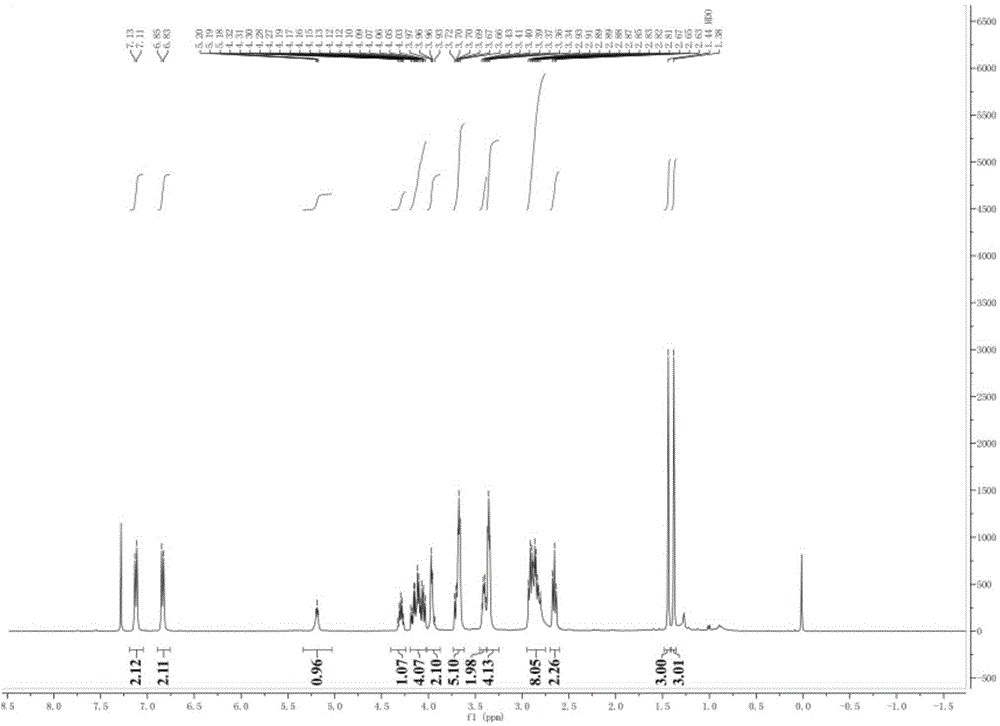
本申請屬於藥物合成
技術領域:
,具體涉及一種鹽酸蘭地洛爾的製備方法。
背景技術:
:鹽酸蘭地洛爾,CAS號:133242-30-5,商品名:Onoact,Onoact50,Onoactforinjection,中文化學名:3-{4-[(2S-羥基-[3-(2-嗎啉甲醯胺基)乙基]氨丙氧基]-苯基}丙酸(2,2-二甲基-1,3-二氧環戊烷-4S)甲基酯鹽酸鹽,英文化學名:(-)-3-[4-2(S)-Hydroxy-3-[2-(morpholinocarboxamido)ethylamino]propoxy]phenyl]propionicacid2,2-dimethyl-1,3-dioxolan-4(S)-ylester。結構式為:鹽酸蘭地洛爾為白色或者類白色塊狀或粉末,分子中有兩個手性中心,四個旋光異構體,臨床上使用的是其單一的S,S構型異構體。鹽酸蘭地洛爾是圍手術期中緊急治療心律失常、手術後動態監測心律失常等不可或缺的急症藥物,自上市後倍受醫生和手術患者的青睞。然而,鹽酸蘭地洛爾的化學結構複雜,製備工藝步驟繁瑣,雜質不易控制,大量製備較為困難,因此目前國內沒有廠家形成規模化生產。但是,鹽酸蘭地洛爾是芳氧丙醇胺類藥物中結構最複雜的化合物,一旦解決其生產工藝問題,具有此類結構的其他40餘種藥物,如倍他洛爾、雷諾嗪、艾司洛爾、氧烯洛爾、美託洛爾等,生產工藝問題都將迎刃而解。因此鹽酸蘭地洛爾生產工藝研究具有非常重要的應用價值。日本專利特開平5-306281公開了一種鹽酸蘭地洛爾合成方法,在該合成方法中,使用了兩個價格特別昂貴的原料:對甲苯磺酸(2,2-二甲基-1,3-二氧戊環-4S)甲基酯及間硝基苯磺酸縮水甘油酯,造成生產成本較高;中間體I、中間體II、蘭地洛爾的純化採用柱層析方法,不利於工業化生產;並且,在製備鹽酸蘭地洛爾之前需要先製備草酸蘭地洛爾,增加了合成步驟,還耗費時間,生產成本也相應增加。US5013734、EP0397031中公開的合成方法也與其類似。中國專利(CN101012217A)公開了一種鹽酸蘭地洛爾合成方法,在該合成方法中,使用S-(+)-環氧氯丙烷為原料,經過優化縮合、酯化、醚化的反應條件,得到3-[4-(2S,3-環氧丙氧基)苯基]丙酸(2,2-二甲基-1,3-二氧戊環-4S)甲基酯;再與使用羰基二咪唑為原料製備的N-(2-氨乙基)嗎啉甲醯胺草酸成鹽在異丙醇水溶液中發生開環反應得到蘭地洛爾,然後通過直接成鹽的方法得到目標產物鹽酸蘭地洛爾。該方法在合成N-(2-氨乙基)-4-嗎啉甲醯胺草酸時存在如下缺陷:(1)反應過程中以氯仿為溶劑,氯仿具有肝毒性,不易生產操作;(2)反應時間長,需要柱層析純化,不宜工業化。王元忠、李勤耕(鹽酸蘭地洛爾的合成工藝研究,重慶醫科大學碩士論文,2014年)等人也提出了鹽酸蘭地洛爾合成方法,在合成中間體式III時,將乙二胺用二碳酸二叔丁酯單邊保護得到2-氨乙基-叔丁氧基甲醯胺,用N,N′-羰基二咪唑和嗎啉反應生成N,N′-羰基嗎啉咪唑,再與2-氨乙基-叔丁氧基甲醯胺反應生成N-(2-氨乙基)嗎啉甲醯胺,用三氟乙酸脫保護,再與草酸成鹽,生成N-(2-氨乙基)嗎啉甲醯胺草酸鹽。在該方法的整個過程中雖然沒有使用柱層析,但製備單BOC保護的乙二胺時需要20當量的乙二胺,原子經濟性很不好。曹珍豔(鹼性紫5、鹽酸蘭地洛爾、三滷異氰尿酸的合成以及拉米夫定的工藝設計,山東大學碩士學位論文,2008年)提出的方法採用對羥基苯丙酸為原料,將羧基轉化成乙酯,然後醚化、開環胺化生成具有芳氧丙醇胺結構的特殊中間體3-{4-[(2S-羥基-[3-(2-嗎啉甲醯氨基)乙基]氨丙氧基]-苯基}丙酸乙酯,最後經皂化、酯交換得到了蘭地洛爾粗品,直接通入HCl得到鹽酸蘭地洛爾。該方法的主要缺點是:(1)使用的原料(2S)-(+)-對甲苯磺酸縮水甘油酯及對甲基苯磺酸(2,2-二甲基-1,3-二氧環戊基-(4S)基)甲基酯非常昂貴;(2)使用了濃硫酸,會腐蝕容器;(3)直接通入氯化氫氣體會產生大量副產物,收率低;(4)整個過程反應步驟過長,終產物產率不高。專利EP2687521A1(或KR2012100823)公開的合成方法為:起始原料是中間體式I和消旋體環氧氯丙烷,第一步以Jacobsen環氧氯丙烷拆分法得到特殊中間體3-{4-[(2R)-2-羥基-3-氯-丙氧基]-苯基}丙酸(2,2-二甲基-1,3-二氧環戊烷-4S)甲基酯;此特殊中間體與中間體式IV經親核取代反應得到蘭地洛爾粗品,然後將蘭地洛爾粗品融入飽和氯化氫異丙醇溶液中即可得到鹽酸蘭地洛爾。該方法原子經濟性好、產率較高,所得到的化合物純度很高,但是第一步反應操作繁瑣,第二步要在回流條件下去除草酸鹽,不易操作。因此,本領域仍需要提供鹽酸蘭地洛爾的新型合成方法。技術實現要素:為解決上述問題,本發明提供一條操作簡便、環境友好、穩定性高、重複性好、適合工業化大生產的新的鹽酸蘭地洛爾合成路線。具體而言,本發明提供鹽酸蘭地洛爾,即3-{4-[(2S-羥基-[3-(2-嗎啉甲醯胺基)乙基]氨丙氧基]-苯基}丙酸(2,2-二甲基-1,3-二氧環戊烷-4S)甲基酯鹽酸鹽的製備方法,所述製備方法包括以下步驟:本發明提供一種鹽酸蘭地洛爾的製備方法,所述製備方法包括以下步驟:(1)在DMSO中加入對羥基苯丙酸、氫氧化鉀、無水碳酸鉀和相轉移催化劑,後滴入(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯,反應生成式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯;(2)將式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯、無水碳酸鉀、相轉移催化劑均溶解於丙酮中,後滴加R-(-)-環氧氯丙烷,反應生成式II所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯;(3)將乙二胺溶解於二氯甲烷,滴加N-咪唑-4-嗎啉甲醯胺的二氯甲烷溶液,後滴加草酸的甲醇溶液,反應生成式III所示N-(2-氨基乙基)-4-嗎啉甲醯胺草酸;(4)向式III所示N-(2-氨基乙基)-4-嗎啉甲醯胺草酸中滴加氫氧化鈉溶液,反應生成式IV所示N-(2-氨基乙基)-4-嗎啉醯胺;(5)將式IV所示N-(2-氨基乙基)-4-嗎啉醯胺溶於水-異丙醇體系中,後滴加式II所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯的異丙醇溶液,反應生成式V所示蘭地洛爾;在式V所示蘭地洛爾中加入飽和氯化銨溶液後滴加鹽酸,反應生成式VI所示鹽酸蘭地洛爾,所述步驟(1)中,所述(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯的製備方法為:將丙酮和S-(+)-環氧氯丙烷混合,滴加三氟化硼乙醚,反應生成(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯;優選地,所述(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯的製備方法為:在氮氣下,將丙酮和S-(+)-環氧氯丙烷混合,控溫-5~5℃,滴加三氟化硼乙醚,後經飽和碳酸氫鈉溶液洗滌,收集上清液,得到(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯;優選地,所述(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯的製備方法為:在氮氣下,將丙酮和S-(+)-環氧氯丙烷混合,冰鹽浴控溫-5~5℃,常壓下滴加三氟化硼乙醚,後升溫至40~45℃下,反應3~6.5小時,後冷卻至室溫,使用飽和碳酸氫鈉溶液洗滌,收集上清液,得到(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯所述步驟(1)為:在DMSO中加入對羥基苯丙酸、氫氧化鉀、無水碳酸鉀、相轉移催化劑,升溫至115~120℃,滴加(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯,反應7~8h,用石油醚和乙酸乙酯的混合溶液進行萃取,後依次用飽和碳酸氫鈉溶液、飽和氯化鈉溶液洗滌,濃縮,得到式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯的粗品;優選地,所述步驟(1)還包括以下步驟:將石油醚與乙酸乙酯的混合溶液加入至式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯的粗品中,脫色,過濾,得到式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯;優選地,所述石油醚和乙酸乙酯的混合溶液中石油醚與乙酸乙酯的體積比為5∶1;優選地,所述步驟(1)中,所述對羥基苯丙酸、氫氧化鉀、無水碳酸鉀、相轉移催化劑的摩爾比為1∶0.98~1.5∶1~2∶0.05~0.2,更優選為1∶0.98∶1.5∶0.1;優選地,所述步驟(1)中,所述對羥基苯丙酸、氫氧化鉀、無水碳酸鉀、相轉移催化劑和(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯的摩爾比為1∶0.98~1.5∶1~2∶0.05~0.2∶1~2.5,更優選為1∶0.98∶1.5∶0.1∶2;優選地,所述步驟(1)中,所述相轉移催化劑選自四正丁基溴化銨、四正丁基碘化銨、三乙基苄基氯化銨或PEG400中的一種或多種,優選為四正丁基溴化銨。所述步驟(2)為:將式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯,以及無水碳酸鉀、相轉移催化劑均溶解於丙酮中,升溫至回流,滴加R-(-)-環氧氯丙烷,反應6~6.5h,過濾,依次用飽和碳酸氫鈉溶液和飽和氯化鈉溶液洗滌,濃縮,得到式II所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯;優選地,所述步驟(2)中,式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯、R-(-)-環氧氯丙烷、無水碳酸鉀和相轉移催化劑的摩爾比為1∶4~5∶2~3∶0.05~0.1,優選為1∶5∶2∶0.05。優選地,所述步驟(2)中,所述相轉移催化劑選自三乙基苄基溴化銨、四正丁基溴化銨、四正丁基碘化銨、四正丁基硫酸氫銨、PEG400中的一種或多種,優選為三乙基苄基溴化銨。所述步驟(3)中,所述N-咪唑-4-嗎啉甲醯胺的製備方法為:將N,N′-羰基二咪唑溶解於二氯甲烷中後,滴加嗎啉的二氯甲烷溶液,反應生成N-咪唑-4-嗎啉甲醯胺;優選地,所述N-咪唑-4-嗎啉甲醯胺的製備方法為:將N,N′-羰基二咪唑溶解在二氯甲烷中後,滴加嗎啉的二氯甲烷溶液,攪拌6~6.5h,水洗滌,二氯甲烷萃取,無水硫酸鈉乾燥,得到N-咪唑-4-嗎啉甲醯胺;優選地,所述N,N′-羰基二咪唑與嗎啉的摩爾比為1∶1。所述步驟(3)為:將乙二胺溶解於二氯甲烷中,滴加N-咪唑-4-嗎啉甲醯胺的二氯甲烷溶液,升溫至回流,減壓,控溫至小於10℃,滴加草酸的甲醇溶液,過濾,濃縮,得到式III所示N-(2-氨基乙基)-4-嗎啉甲醯胺草酸;優選地,所述步驟(3)為:將乙二胺溶解在二氯甲烷中,攪拌,滴加N-咪唑-4-嗎啉甲醯胺的二氯甲烷溶液,升溫至回流,反應34~38h,減壓將二氯甲烷和剩餘的乙二胺蒸出後加入二氯甲烷,繼續減壓蒸餾至無餾分為止,使用甲醇溶解濃縮殘留物,控溫至小於10℃,滴加草酸的甲醇溶液,直至pH值為4~5時,過濾,濃縮,加入乙酸乙酯結晶,得到式III所示N-(2-氨基乙基)-4-嗎啉甲醯胺草酸;優選地,所述步驟(3)中,所述N-咪唑-4-嗎啉甲醯胺與所述乙二胺的摩爾比為1∶6。所述步驟(4)為:將式III所示N-(2-氨基乙基)-4-嗎啉甲醯胺草酸溶於水,43~47℃下滴加氫氧化鈉溶液,過濾,濃縮,得到式IV所示N-(2-氨基乙基)-4-嗎啉醯胺;優選地,所述步驟(4)為:將式III所示N-(2-氨基乙基)-4-嗎啉甲醯胺草酸溶於水,43~47℃下滴加40%氫氧化鈉溶液,調節pH至10~11.5,攪拌20~30min,過濾,濃縮,加入丙酮溶解,加入無水硫酸鈉乾燥,過濾,濃縮,得到式IV所示N-(2-氨基乙基)-4-嗎啉醯胺。所述步驟(5)為:將式IV所示N-(2-氨基乙基)-4-嗎啉醯胺溶於水-異丙醇體系中,40~60℃下,滴加式II所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯的異丙醇溶液,反應4~6h,得到式V所示蘭地洛爾;在式V所示蘭地洛爾中加入飽和氯化銨溶液,10℃以下滴加鹽酸使體系的pH為4.5~5,乾燥,濃縮,得到式VI所示鹽酸蘭地洛爾;優選地,所述步驟(5)為:將式IV所示N-(2-氨基乙基)-4-嗎啉醯胺溶於水-異丙醇體系中,40~60℃下,滴加式II所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯的異丙醇溶液,反應4~6h,過濾,濃縮,加入飽和氯化鈉溶液至全溶,萃取,洗滌,乾燥,過濾,濃縮,加入乙酸乙酯溶解,脫色,得到式V所示蘭地洛爾;在式V所示蘭地洛爾中加入飽和氯化銨溶液,分液,10℃以下滴加0.1~0.3N鹽酸使體系的pH為4.5~5,加入氯化銨至飽和,加入乙酸乙酯萃取,無水硫酸鈉乾燥,濃縮,加入乙酸乙酯,析出絮狀固體,過濾,衝洗,真空乾燥,得到式VI所示鹽酸蘭地洛爾;優選地,所述步驟(5)中,所述式II所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯與式IV所示N-(2-氨基乙基)-4-嗎啉醯胺的摩爾比為1∶2~2.5;優選為1∶2.5;優選地,所述步驟(5)中,所述水-異丙醇體系中,所述水和異丙醇的體積比為1∶5~6;優選為1∶6。所述製備方法包括以下步驟:(1)在0~25℃和氮氣保護下,加入丙酮和S-(+)-環氧氯丙烷,攪拌,冰鹽浴控溫-5~5℃,常壓下滴加三氟化硼乙醚,攪拌,升溫至40~45℃反應3~6.5小時,冷卻至室溫,飽和碳酸氫鈉洗滌,收集上清液,得到(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯;室溫下,在DMSO中加入摩爾比為1∶0.98∶1.5∶0.1的對羥基苯丙酸、氫氧化鉀、無水碳酸鉀、四正丁基溴化銨,升溫至115~120℃,攪拌,滴加上述得到的(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯,TLC監控下,反應7~8h,使用體積比為5~6∶1的石油醚和乙酸乙酯的混合溶液萃取3次,合併萃取液,依次用飽和碳酸氫鈉溶液、飽和氯化鈉溶液洗滌兩次,經無水硫酸鈉乾燥、減壓濃縮得到式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯粗品;將體積比為5∶1的石油醚與乙酸乙酯的混合溶液加入上述得到的粗品,升溫溶解,加入活性炭,脫色,過濾,結晶,真空乾燥,得到式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯;其中,所述對羥基苯丙酸、氫氧化鉀、無水碳酸鉀、相轉移催化劑和(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯的摩爾比為1∶0.98∶1.5∶0.1∶2;(2)取摩爾比為1∶5∶2∶0.05的式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯、無水碳酸鉀、三乙基苄基溴化銨和丙酮,將式I所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯、無水碳酸鉀、三乙基苄基溴化銨溶解於丙酮中,升溫至回流,攪拌0.5h,滴加R-(-)-環氧氯丙烷,反應6~6.5h,過濾,濃縮,後用乙酸乙酯溶解,依次用飽和碳酸氫鈉溶液和飽和氯化鈉溶液洗滌2次,無水硫酸鈉乾燥,過濾,減壓,濃縮,得到式II所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯;(3)將N,N′-羰基二咪唑溶解於二氯甲烷中,攪拌,恆壓滴加嗎啉的二氯甲烷溶液,其中,所述N,N′-羰基二咪唑與嗎啉的摩爾比為1∶1,滴加完成後室溫攪拌6~6.5h,經水洗滌,二氯甲烷萃取,無水硫酸鈉乾燥,得到N-咪唑-4-嗎啉甲醯胺;室溫下,將乙二胺溶解在二氯甲烷溶液中,攪拌,恆壓滴加N-咪唑-4-嗎啉甲醯胺的二氯甲烷溶液,其中所述N-咪唑-4-嗎啉甲醯胺與所述乙二胺的摩爾比為1∶6,升溫至回流,反應34~38h,減壓蒸出二氯甲烷和剩餘的乙二胺後加入二氯甲烷,繼續減壓蒸餾至無餾分為止,使用甲醇溶解濃縮殘留物,冰浴控溫小於10℃,緩慢滴加草酸的甲醇溶液,直至pH值為4~5時停止滴加,過濾,濃縮,加入乙酸乙酯結晶,得到式III所示N-(2-氨基乙基)-4-嗎啉甲醯胺草酸;(4)將式III所示N-(2-氨基乙基)-4-嗎啉甲醯胺草酸溶於水,43~47℃下滴加40%的氫氧化鈉溶液,調節pH至10~11.5,保溫,繼續攪拌20~30min,過濾,濃縮,濃縮物中加入丙酮溶解,無水硫酸鈉乾燥,過濾,濃縮得到式IV所示N-(2-氨基乙基)-4-嗎啉醯胺;(5)將式IV所示N-(2-氨基乙基)-4-嗎啉醯胺溶於體積比為1∶6的水-異丙醇體系中,在40~60℃的條件下,滴加式II所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯的異丙醇溶液,其中式II所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯與式IV所示N-(2-氨基乙基)-4-嗎啉醯胺的摩爾比為1∶2.5,滴加完畢後繼續反應4~6h,過濾,濃縮,加入飽和氯化鈉溶液至全溶,經乙酸乙酯萃取,經飽和氯化鈉溶液洗滌和無水硫酸鈉乾燥,過濾,濃縮,後加入乙酸乙酯溶解,加入活性炭,加熱回流脫色5min,過濾濃縮,得到式V所示蘭地洛爾;將式V所示蘭地洛爾加入飽和氯化銨溶液中,靜置1h,分液,除去油層後,將剩餘體系在10℃以下滴加0.1~0.3N鹽酸至pH為4.5~5,加入氯化銨至飽和,加入乙酸乙酯萃取3次,合併萃取液,經無水硫酸鈉乾燥,濃縮,後加入乙酸乙酯,加熱回流,過濾,降溫,析出固體,過濾,乙酸乙酯衝洗三次,真空乾燥,得到式VI所示鹽酸蘭地洛爾。具體來說,本發明包括如下步驟:(1)中間體式I3-(4-羥基苯基)丙酸-[(S)-(2,2-二甲基-1,3-二氧環戊烷基)-4-]甲酯的合成:第一步,合成(S)-2,2-二甲基-1,3-二氧環戊烷-4-甲基氯:控溫0~25℃,在氮氣的保護下,加入丙酮和S-(+)-環氧氯丙烷,攪拌,冰鹽浴控溫-5~5℃,常壓下緩慢滴加三氟化硼乙醚,繼續攪拌,然後再40~45℃下反應3~6.5小時,停止攪拌,冷卻至室溫,飽和碳酸氫鈉洗滌,收集上清液,得到(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯;第二步,合成中間體式I:室溫下,在DMSO中加入摩爾比為1∶0.98∶1.5∶0.1的對羥基苯丙酸、氫氧化鉀、無水碳酸鉀、四正丁基溴化銨,升溫至115~120℃,攪拌,滴加與對羥基苯丙酸的摩爾比為1.5~2∶1的(S)-2,2-二甲基-1,3-二氧環戊烷-4-甲基氯,TLC監控反應進度,繼續反應7~8h,用石油醚和乙酸乙酯的混合溶液萃取3次,合併萃取液,依次用飽和碳酸氫鈉、飽和氯化鈉溶液洗滌,經無水硫酸鈉乾燥、減壓濃縮得到中間體I粗品;用5∶1的石油醚與乙酸乙酯的混合液重結晶,真空乾燥,得白色針狀結晶中間體I;其中,所述對羥基苯丙酸、氫氧化鉀、無水碳酸鉀、相轉移催化劑和(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯的摩爾比為1∶0.98∶1.5∶0.1∶2。(2)中間體式II3-(4-(S)-環氧丙烷基甲氧基)苯丙酸-[(S)-(2,2-二甲基-1,3-二氧環戊烷基)-4-]甲酯的合成:將摩爾比為1∶5∶2∶0.05的中間體式I、無水碳酸鉀、三乙基苄基溴化銨溶解在丙酮中,升溫至回流,攪拌0.5h,滴加R-(-)-環氧氯丙烷,反應6~6.5h,過濾,濃縮,殘留物用乙酸乙酯溶解,依次用飽和碳酸氫鈉和飽和氯化鈉洗滌2次,無水硫酸鈉乾燥,過濾,減壓濃縮,即可得到中間體式II;(3)中間體式IIIN-(2-氨基乙基)-4-嗎啉甲醯胺草酸的合成:第一步,合成N-咪唑-4-嗎啉甲醯胺:將N,N′-羰基二咪唑溶解在二氯甲烷中,攪拌,滴加嗎啉的二氯甲烷溶液,其中,所述N,N′-羰基二咪唑與嗎啉的摩爾比為1∶1,滴完後室溫攪拌6~6.5h,再經水洗滌,二氯甲烷萃取,無水硫酸鈉乾燥等步驟,得到N-咪唑-4-嗎啉甲醯胺;第二步,合成中間體式III:將乙二胺溶解在二氯甲烷中,攪拌,室溫下滴加N-咪唑-4-嗎啉甲醯胺的二氯甲烷溶液,其中所述N-咪唑-4-嗎啉甲醯胺與所述乙二胺的摩爾比為1∶6,;然後,升溫至回流反應35h,減壓蒸出二氯甲烷和剩餘的乙二胺,剩餘物用甲醇溶解,在<10℃下緩慢滴加草酸的甲醇溶液,調節pH值為4~5,過濾濃縮,加入乙酸乙酯結晶,得到中間體式III;(4)中間體式IVN-(2-氨基乙基)-4-嗎啉醯胺的合成將中間體式III溶於水中,在43~47℃的條件下滴加40%的氫氧化鈉溶液,調節pH至10~11.5,保溫繼續攪拌20min,過濾,濃縮,濃縮物加入丙酮溶解,無水硫酸鈉乾燥,過濾濃縮得到中間體式IV;(5)式V蘭地洛爾的合成將中間體式IV溶於體積比為1∶6的水-異丙醇體系中,在40~60℃的條件下,緩慢滴加中間體式II的異丙醇溶液,其中中間體式II與中間體式IV的摩爾比為1∶2~2.5,其中式II所示4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯與式IV所示N-(2-氨基乙基)-4-嗎啉醯胺的摩爾比為1∶2.5,滴加完畢後繼續反應4~6h,過濾濃縮,加入飽和氯化鈉至全溶,乙酸乙酯萃取,飽和氯化鈉溶液洗滌、無水硫酸鈉乾燥,過濾濃縮,再加入乙酸乙酯溶解,活性炭脫色,過濾濃縮,得到式V蘭地洛爾;(6)式VI鹽酸蘭地洛爾的合成將式V所示蘭地洛爾加入飽和氯化銨溶液中,轉移到分液漏鬥中,靜置1h,分液,除去油層,剩餘體系在10℃以下緩慢滴加0.1~0.3N鹽酸使體系的pH為4.5~5,加入氯化鈉使體系達到飽和,用乙酸乙酯萃取3次,合併萃取液,無水硫酸鈉乾燥,濃縮得灰白色粗品;再加入乙酸乙酯,加熱回流至全部溶解,趁熱過濾,緩慢降溫,析出絮狀固體,過濾,乙酸乙酯衝洗三次,真空乾燥,即得鹽酸蘭地洛爾。本發明方法的合成路線如下:與現有技術相比,本發明具有如下優點:(1)原料便宜易獲得;(2)關鍵反應使用相轉移催化法,縮短了反應時間;(3)簡化了操作,提高了收率;(4)避免了不適於工業化生產的柱層析純化操作,採用飽和氯化銨/稀鹽酸溶液成鹽法產品質量容易控制。附圖說明圖1為本發明所述鹽酸蘭地洛爾H1-NMR;圖2為本發明所述鹽酸蘭地洛爾C13-NMR。具體實施方式以下參照具體的實施例來說明本發明。本領域技術人員能夠理解,這些實施例僅用於說明本發明,其不以任何方式限制本發明的範圍。下述實施例中的實驗方法,如無特殊說明,均為常規方法。下述實施例中所用的藥材原料、試劑材料等,如無特殊說明,均為市售購買產品。實施例1:(1)式I,即4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯的合成:控溫20℃,氮氣保護下,將丙酮58.08g和(S)-(+)-環氧氯丙烷18.50g加入到乾燥的250mL三口瓶中,攪拌十分鐘;冰鹽浴控溫0℃,常壓下緩慢滴加1.42g三氟化硼乙醚,繼續控溫攪拌十分鐘後升溫至40℃,體系逐漸變黃,6小時後完成反應,停止攪拌,冷卻至室溫;將飽和NaHCO3(40mL)溶液緩慢加入反應瓶中,淬滅反應,體系冒泡,逐漸變澄清,收集上清液,上清液中再加入飽和NaHCO3(40mL)洗滌;收集上清液,濃縮得到無色透明油狀物,得到(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯28.6g;室溫下,在40mLDMSO中依次加入3-(4-羥基苯基)丙酸10.0g、氫氧化鉀3.3g、無水碳酸鉀12.40g、四丁基溴化銨1.93g,將上述物置於100mL三口瓶中,升溫至115℃,攪拌0.5h,體系變黑,滴加(2,2-二甲基-1,3-二氧戊環-4S)甲基氯18.07g,TLC監控反應進度,繼續反應7~8小時;反應完成後,過濾,收集濾液,用石油醚(v)∶乙酸乙酯(v)=5∶1(50mL)共三次提取產物,合併提取液,依次用飽和NaHCO3溶液(110mL×2)、飽和NaCl溶液洗滌(110mL×2)兩次,無水硫酸鈉乾燥(6g)1小時;過濾,減壓濃縮溶劑得到中間體式I的粗品(淡黃色)。加入石油醚(v)∶乙酸乙酯(v)=6∶1(56mL)至中間體式I的粗品中,升溫沸騰使中間體式I粗品完全溶解,加入活性炭1g,脫色3分鐘,趁熱過濾,濾液冷卻析出白色針狀結晶,過濾,收集濾餅,真空乾燥(35℃下)得中間體式I14.6g,收率為85.62%。(2)式II,即4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯的合成:中間體式I5.60g、無水碳酸鉀5.53g、三乙基丁基溴化銨272mg加入到100mL三口瓶中,加入溶劑丙酮45mL,升溫至回流,攪拌0.5h,滴加(R)-(-)-環氧氯丙烷9.25g,TLC監測反應進度,繼續反應6~6.5小時;反應完成後冷卻至室溫,過濾,濾餅用少量丙酮衝洗(3mL×3),濾液濃縮,殘留物用乙酸乙酯溶解,依次用飽和NaHCO3溶液(40mL×1)、飽和NaCl洗滌溶液(40mL×2)洗滌,3g無水硫酸鈉乾燥3小時;過濾,減壓濃縮溶劑的得到中間體式II所示化合物,淡黃色油狀物6.26g,收率為93.0%。(3)式III,即N-(2-氨基乙基)-4-嗎啉甲醯胺草酸的合成合成N-咪唑-4-嗎啉甲醯胺:室溫,將CDI16.21g及80mL二氯甲烷加入250mL三口瓶中,攪拌,恆壓滴加嗎啉8.7g的二氯甲烷(50mL)溶液,1小時左右滴完,室溫繼續反應5~6小時;反應完成後加入60mL水洗滌,水層再用二氯甲烷萃取(50mLx3),合併有機層,加入5g無水硫酸鈉乾燥3小時,過濾,濃縮得到白色固體N-咪唑-4-嗎啉甲醯胺16.42g,收率為90.62%;合成式III所示化合物:室溫,將乙二胺18.03g及50mL二氯甲烷加入250mL三口瓶中,攪拌,恆壓緩慢滴加含9.1gN-咪唑-4-嗎啉甲醯胺的二氯甲烷(100mL)溶液,升溫至回流,反應36h左右;反應完成後室溫減壓蒸出二氯甲烷,65℃,減壓至P≤-0.07Mpa,蒸出剩餘的乙二胺,至無餾分為止,剩餘體系加入20mL甲醇,繼續65℃,減壓至P≤-0.07Mpa蒸餾,至無餾分為止,剩餘體系加入20mL二氯甲烷,繼續65℃減壓蒸餾,至無餾分為止;最後用甲醇50mL溶解,冰浴控溫小於10℃,緩慢滴加草酸的甲醇溶液,直至pH值為4~5時停止滴加,過濾,濾液濃縮至無餾分,加入乙酸乙酯80mL過夜,析出白色固體,過濾,收集濾餅,真空乾燥(35℃下)得中間體式III10.56g,收率為80.23%;(4)式IV,即N-(2-氨基乙基)-4-嗎啉醯胺的合成:室溫條件下,在100mL反應瓶中加入式III所示化合物15.8g和水47.4g,攪拌,升溫至43~47℃全溶,攪拌滴加40%氫氧化鈉溶液,調節pH至10~11.5,保溫繼續攪拌20分鐘左右;冷卻至室溫,過濾,濾餅用7.2g水衝洗,濾液轉入旋蒸瓶,於44~48℃、壓力P≤-0.07Mpa下,濃縮至無餾分餾出,剩餘殘留物降至室溫,加入丙酮110g,攪拌至殘留物全部分散於丙酮中,後轉入250mL的三角瓶中,加無水硫酸鈉9.48g乾燥3小時,過濾,濾液轉入茄形旋蒸瓶濃縮至無餾分,得到中間體式IV純品,淡黃色油狀物8.8g,收率為84.67%。(5)式VI,即鹽酸蘭地洛爾的合成將8.6g式IV所示化合物、4mL水、20mL異丙醇加入至100mL三口燒瓶中,攪拌升溫至50~55℃,緩慢滴加6.7g式II所示化合物的異丙醇溶液(異丙醇溶液為4mL),滴加完畢後,保溫50℃繼續反應5~5.5小時;反應完成後小於45℃濃縮,至無餾分,剩餘體系加入25mL飽和氯化鈉,攪拌至全溶,十分鐘後,加入25mL乙酸乙酯,繼續攪拌10min,萃取,分液,收集有機層,重複三次,合併有機層,有機層中加入飽和氯化鈉(50mL)洗滌2次,加入5g無水硫酸鈉乾燥,過濾,濃縮至無餾分,再次加入30mL乙酸乙酯溶解,加入1g活性炭,加熱回流,脫色5min左右(或常溫脫色1h),趁熱過濾,濾液冷卻至室溫,濃縮得到式V所示蘭地洛爾的粗品,黃色油狀物9.4g,收率為92.30%。將式V所示蘭地洛爾的粗品5.09g加入到100mL茄形燒瓶中,加入飽和氯化銨30mL攪拌1h,轉移到125mL的分液漏鬥中,靜置1h左右,分液,除去壁上的油層;剩餘體系轉移到100mL潔淨的茄形燒瓶中,冰水浴下,控溫10℃以下,緩慢滴加0.2N鹽酸使體系的pH至4.5~5左右,加入少量的氯化鈉或者氯化銨使體系重新達到飽和,加入30mL乙酸乙酯萃取3次,合併乙酸乙酯層後加入6g無水硫酸鈉乾燥,濃縮得灰白色殘渣,將35mL的乙酸乙酯加入殘渣中,加熱回流至白色殘渣全部溶解,趁熱過濾轉移至新的100mL茄形瓶中,緩慢降溫,析出絮狀固體,過濾,用5mL乙酸乙酯衝洗3次。將濾餅放入35℃真空乾燥箱中乾燥至恆重,得式VI所示鹽酸蘭地洛爾4.60g,收率為84.29%,純度為99.6%。1HNMR(400MHz,CDCl3):δ7.13-7.11(d,J=8.4Hz,1H),6.85-6.83(d,J=8.3Hz,1H),5.19(s,1H),4.32-4.27(m,1H),4.19-4.03(m,4H),3.97-3.96(d,J=4.2Hz,2H),3.72-3.66(m,5H),3.41-3.34(m,6H),2.87-2.82(ddd,J=16.5,13.1,5.6Hz,8H),2.67-2.63(t,J=7.7Hz,2H),1.44(s,3H),1.38(s,3H).13CNMR(100MHz,CDCl3):δ173.71,159.08,157.54,134.32,129.35,114.57,109.84,73.57,70.38,68.37,66.45,66.32,64.73,51.47,49.29,43.97,40.69,35.89,30.00,26.69,25.39.實施例2:(1)式I,即4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯的合成:控溫20℃,氮氣保護下,將丙酮58.08g和(S)-(+)-環氧氯丙烷18.50g加入到乾燥的250mL三口瓶中,攪拌十分鐘;冰鹽浴控溫0℃,常壓下緩慢滴加1.42g三氟化硼乙醚,繼續控溫攪拌十分鐘後升溫至45℃,體系逐漸變黃,3小時後完成反應,停止攪拌,冷卻至室溫;將飽和NaHCO3(40mL)溶液緩慢加入反應瓶中,淬滅反應,體系冒泡,逐漸變澄清,收集上清液,上清液中再加入飽和NaHCO3(40mL)洗滌;收集上清液,濃縮得到無色透明油狀物,得到(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯25.49g;室溫下,在40mLDMSO中依次將3-(4-羥基苯基)丙酸10.0g、氫氧化鉀3.3g、無水碳酸鉀16.58g、TBAB967mg加入到100mL三口瓶中,升溫至120℃,攪拌0.5h,體系變黑,滴加(2,2-二甲基-1,3-二氧戊環-4S)甲基氯18.07g,TLC監控反應進度,繼續反應7~8小時;反應完成後,過濾,收集濾液,用石油醚(v)∶乙酸乙酯(v)=5∶1(50mL)共三次提取產物,合併提取液,依次用飽和NaHCO3溶液(110mL×2)、飽和NaCl溶液洗滌(110mL×2)兩次,無水硫酸鈉乾燥(6g)1小時;過濾,減壓濃縮溶劑得到中間體式I的粗品(淡黃色)。加入石油醚(v)∶乙酸乙酯(v)=5∶1(56mL)至中間體式I的粗品中,升溫沸騰使中間體式I粗品完全溶解,加入活性炭1g,脫色3分鐘,趁熱過濾,濾液冷卻析出白色針狀結晶,過濾,收集濾餅,真空乾燥(35℃下)得中間體式I11.93g,收率為70%。(2)式II,即4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯的合成:中間體式I5.60g、無水碳酸鉀5.53g、TEBAB544mg加入到100mL三口瓶中,加入溶劑丙酮45mL,升溫至回流,攪拌0.5h,滴加(R)-(-)-環氧氯丙烷7.4g,TLC監測反應進度,繼續反應6~6.5小時;反應完成後冷卻至室溫,過濾,濾餅用少量丙酮衝洗(3mL×3),濾液濃縮,殘留物用乙酸乙酯溶解,依次用飽和NaHCO3溶液(40mL×1)、飽和NaCl洗滌溶液(40mL×2)洗滌,3g無水硫酸鈉乾燥3小時;過濾,減壓濃縮溶劑的得到中間體式II所示化合物,淡黃色油狀物5.72g,收率為85.0%。(3)式III,即N-(2-氨基乙基)-4-嗎啉甲醯胺草酸的合成合成N-咪唑-4-嗎啉甲醯胺:室溫,將CDI16.21g及80mL二氯甲烷加入250mL三口瓶中,攪拌,恆壓滴加嗎啉8.7g的二氯甲烷(50mL)溶液,1小時左右滴完,室溫繼續反應5~6小時;反應完成後加入60mL水洗滌,水層再用二氯甲烷萃取(50mLx3),合併有機層,加入5g無水硫酸鈉乾燥3小時,過濾,濃縮得到白色固體N-咪唑-4-嗎啉甲醯胺16.42g,收率為90.62%;合成式III所示化合物:室溫,將乙二胺15.03g及50mL二氯甲烷加入250mL三口瓶中,攪拌,恆壓緩慢滴加含9.1gN-咪唑-4-嗎啉甲醯胺的二氯甲烷(100mL)溶液,升溫至回流,反應34h左右;反應完成後室溫減壓蒸出二氯甲烷,65℃,P≤-0.07Mpa減壓蒸出剩餘的乙二胺,至無餾分為止,剩餘體系加入20mL甲醇,繼續65℃,P≤-0.07Mpa減壓蒸餾,至無餾分為止,剩餘體系加入20mL二氯甲烷,繼續65℃減壓蒸餾,至無餾分為止;最後用甲醇50mL溶解,冰浴控溫小於10℃,緩慢滴加草酸的甲醇溶液,直至pH值為4~5時停止滴加,過濾,濾液濃縮至無餾分,加入乙酸乙酯80mL過夜,析出白色固體,過濾,收集濾餅,真空乾燥(35℃下)得中間體式III9.50g,收率為72.20%;(4)式IV,即N-(2-氨基乙基)-4-嗎啉醯胺的合成:室溫條件下,在100mL反應瓶中加入式III所示化合物15.8g和水47.4g,攪拌,升溫至43~47℃下攪拌滴加40%氫氧化鈉溶液,調節pH至10~11.5,保溫繼續攪拌25分鐘左右;冷卻至室溫,過濾,濾餅用7.2g水衝洗,濾液轉入旋蒸瓶,於44~48℃、P≤-0.07Mpa下,濃縮至無餾分餾出,剩餘殘留物降至室溫,加入丙酮110g,攪拌至殘留物全部分散於丙酮中,後轉入250mL的三角瓶中,加無水硫酸鈉9.48g乾燥3小時,過濾,濾液轉入茄形旋蒸瓶濃縮至無餾分,得到中間體式IV純品,淡黃色油狀物8.8g,收率為84.67%。(5)式VI,即鹽酸蘭地洛爾的合成將6.93g式IV所示化合物、4mL水、18mL異丙醇加入100mL的三口燒瓶中,攪拌升溫至50~55℃,緩慢滴加6.7g式II所示化合物的異丙醇溶液(異丙醇液4mL),滴加完畢後,保溫50℃繼續反應5~5.5小時;反應完成後小於45℃濃縮,至無餾分,剩餘體系加入25mL飽和氯化鈉,攪拌至全溶,十分鐘後,加入25mL乙酸乙酯,繼續攪拌10min,萃取,分液,收集有機層,重複三次,合併有機層,有機層中加入飽和氯化鈉(50mL)洗滌2次,加入5g無水硫酸鈉乾燥,過濾,濃縮至無餾分,再次加入30mL乙酸乙酯溶解,加入1g活性炭,加熱回流,脫色5min左右(或常溫脫色1h),趁熱過濾,濾液冷卻至室溫,濃縮得到式V所示蘭地洛爾的粗品,黃色油狀物9.07g,收率為89.04%。將式V所示蘭地洛爾的粗品5.09g加入到100mL茄形燒瓶中,加入飽和氯化銨30mL攪拌1h,轉移到125mL的分液漏鬥中,靜置1h左右,分液,除去壁上的油層;剩餘體系轉移到100mL潔淨的茄形燒瓶中,冰水浴下,控溫10℃以下,緩慢滴加0.2N鹽酸使體系的pH至4.5~5左右,加入少量的氯化鈉或者氯化銨使體系重新達到飽和,加入30mL乙酸乙酯萃取3次,合併乙酸乙酯層後加入6g無水硫酸鈉乾燥,濃縮得灰白色殘渣,將35mL的乙酸乙酯加入殘渣中,加熱回流至白色殘渣全部溶解,趁熱過濾轉移至新的100mL茄形瓶中,緩慢降溫,析出絮狀固體,過濾,用5mL乙酸乙酯衝洗3次。將濾餅放入35℃真空乾燥箱中乾燥至恆重,得式VI所示鹽酸蘭地洛爾4.58g,收率為83.88%,純度為99.6%。1HNMR(400MHz,CDCl3):δ7.13-7.11(d,J=8.4Hz,1H),6.85-6.83(d,J=8.3Hz,1H),5.19(s,1H),4.32-4.27(m,1H),4.19-4.03(m,4H),3.97-3.96(d,J=4.2Hz,2H),3.72-3.66(m,5H),3.41-3.34(m,6H),2.87-2.82(ddd,J=16.5,13.1,5.6Hz,8H),2.67-2.63(t,J=7.7Hz,2H),1.44(s,3H),1.38(s,3H).13CNMR(100MHz,CDCl3):δ173.71,159.08,157.54,134.32,129.35,114.57,109.84,73.57,70.38,68.37,66.45,66.32,64.73,51.47,49.29,43.97,40.69,35.89,30.00,26.69,25.39.實施例3:(1)式I,即4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯的合成:控溫20℃,氮氣保護下,將丙酮58.08g和(S)-(+)-環氧氯丙烷18.50g加入到乾燥的250mL三口瓶中,攪拌十分鐘;冰鹽浴控溫0℃,常壓下緩慢滴加1.42g三氟化硼乙醚,繼續控溫攪拌十分鐘後升溫至40℃,體系逐漸變黃,4小時後完成反應,停止攪拌,冷卻至室溫;將飽和NaHCO3(40mL)溶液緩慢加入反應瓶中,淬滅反應,體系冒泡,逐漸變澄清,收集上清液,上清液中再加入飽和NaHCO3(40mL)洗滌;收集上清液,濃縮得到無色透明油狀物,得到(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯27.35g;室溫下,在40mLDMSO中依次將3-(4-羥基苯基)丙酸10.0g、氫氧化鉀3.3g、無水碳酸鉀12.40g、TBAB967mg加入到100mL三口瓶中,升溫至115℃,攪拌0.5h,體系變黑,滴加(2,2-二甲基-1,3-二氧戊環-4S)甲基氯9.04g,TLC監控反應進度,繼續反應7~8小時;反應完成後,過濾,收集濾液,用石油醚(v)∶乙酸乙酯(v)=5∶1(50mL)共三次提取產物,合併提取液,依次用飽和NaHCO3溶液(110mL×2)、飽和NaCl溶液洗滌(110mL×2)兩次,無水硫酸鈉乾燥(6g)1小時;過濾,減壓濃縮溶劑得到中間體式I的粗品(淡黃色)。加入石油醚(v)∶乙酸乙酯(v)=5∶1(56mL)至中間體式I的粗品中,升溫沸騰使中間體式I粗品完全溶解,加入活性炭1g,脫色3分鐘,趁熱過濾,濾液冷卻析出白色針狀結晶,過濾,收集濾餅,真空乾燥(35℃下)得中間體式I6.82g,收率為40%。(2)式II,即4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯的合成:中間體式I5.60g、無水碳酸鉀8.29g、TEBAB272mg加入到100mL三口瓶中,加入溶劑丙酮45mL,升溫至回流,攪拌0.5h,滴加(R)-(-)-環氧氯丙烷9.25g,TLC監測反應進度,繼續反應6~6.5小時;反應完成後冷卻至室溫,過濾,濾餅用少量丙酮衝洗(3mL×3),濾液濃縮,殘留物用乙酸乙酯溶解,依次用飽和NaHCO3溶液(40mL×1)、飽和NaCl洗滌溶液(40mL×2)洗滌,3g無水硫酸鈉乾燥3小時;過濾,減壓濃縮溶劑的得到中間體式II所示化合物,淡黃色油狀物4.71g,收率為70.0%。(3)式III,即N-(2-氨基乙基)-4-嗎啉甲醯胺草酸的合成合成N-咪唑-4-嗎啉甲醯胺:室溫,將CDI16.21g及80mL二氯甲烷加入250mL三口瓶中,攪拌,恆壓滴加嗎啉10.44g的二氯甲烷(50mL)溶液,1小時左右滴完,室溫繼續反應5~6小時;反應完成後加入60mL水洗滌,水層再用二氯甲烷萃取(50mLx3),合併有機層,加入5g無水硫酸鈉乾燥3小時,過濾,濃縮得到白色固體N-咪唑-4-嗎啉甲醯胺16.33g,收率為90.12%;合成式III所示化合物:室溫,將乙二胺18.03g及50mL二氯甲烷加入250mL三口瓶中,攪拌,恆壓緩慢滴加含9.1gN-咪唑-4-嗎啉甲醯胺的二氯甲烷(100mL)溶液,升溫至回流,反應38h左右;反應完成後室溫減壓蒸出二氯甲烷,65℃,P≤-0.07Mpa減壓蒸出剩餘的乙二胺,至無餾分為止,剩餘體系加入20mL甲醇,繼續65℃,P≤-0.07Mpa減壓蒸餾,至無餾分為止,剩餘體系加入20mL二氯甲烷,繼續65℃減壓蒸餾,至無餾分為止;最後用甲醇50mL溶解,冰浴控溫小於10℃,緩慢滴加草酸的甲醇溶液,直至pH值為4~5時停止滴加,過濾,濾液濃縮至無餾分,加入乙酸乙酯80mL過夜,析出白色固體,過濾,收集濾餅,真空乾燥(35℃下)得中間體式III10.53g,收率為80.0%;(4)式IV,即N-(2-氨基乙基)-4-嗎啉醯胺的合成:室溫條件下,在100mL反應瓶中加入式III所示化合物15.8g和水47.4g,攪拌,升溫至43~47℃下攪拌滴加40%氫氧化鈉溶液,調節pH至10~11.5,保溫繼續攪拌30分鐘左右;冷卻至室溫,過濾,濾餅用7.2g水衝洗,濾液轉入旋蒸瓶,於44~48℃、P≤-0.07Mpa下,濃縮至無餾分餾出,剩餘殘留物降至室溫,加入丙酮110g,攪拌至殘留物全部分散於丙酮中,後轉入250mL的三角瓶中,加無水硫酸鈉9.48g乾燥3小時,過濾,濾液轉入茄形旋蒸瓶濃縮至無餾分,得到中間體式IV純品,淡黃色油狀物8.5g,收率為81.78%。(5)式VI,即鹽酸蘭地洛爾的合成將8.6g式IV所示化合物、4mL水、20mL異丙醇加入100mL的三口燒瓶中,攪拌升溫至40~45℃,緩慢滴加6.7g式II所示化合物的異丙醇溶液(異丙醇液4mL),滴加完畢後,保溫50℃繼續反應5~5.5小時;反應完成後小於45℃濃縮,至無餾分,剩餘體系加入25mL飽和氯化鈉,攪拌至全溶,十分鐘後,加入25mL乙酸乙酯,繼續攪拌10min,萃取,分液,收集有機層,重複三次,合併有機層,有機層中加入飽和氯化鈉(50mL)洗滌2次,加入5g無水硫酸鈉乾燥,過濾,濃縮至無餾分,再次加入30mL乙酸乙酯溶解,加入1g活性炭,加熱回流,脫色5min左右(或常溫脫色1h),趁熱過濾,濾液冷卻至室溫,濃縮得到式V所示蘭地洛爾的粗品,黃色油狀物8.64g,收率為84.87%。將式V所示蘭地洛爾的粗品5.09g加入到100mL茄形燒瓶中,加入飽和氯化銨30mL攪拌1h,轉移到125mL的分液漏鬥中,靜置1h左右,分液,除去壁上的油層;剩餘體系轉移到100mL潔淨的茄形燒瓶中,冰水浴下,控溫10℃以下,緩慢滴加0.2N鹽酸使體系的pH至4.5~5左右,加入少量的氯化鈉或者氯化銨使體系重新達到飽和,加入30mL乙酸乙酯萃取3次,合併乙酸乙酯層後加入6g無水硫酸鈉乾燥,濃縮得灰白色殘渣,將35mL的乙酸乙酯加入殘渣中,加熱回流至白色殘渣全部溶解,趁熱過濾轉移至新的100mL茄形瓶中,緩慢降溫,析出絮狀固體,過濾,用5mL乙酸乙酯衝洗3次。將濾餅放入35℃真空乾燥箱中乾燥至恆重,得式VI所示鹽酸蘭地洛爾4.54g,收率為83.14%,純度為99.6%。1HNMR(400MHz,CDCl3):δ7.13-7.11(d,J=8.4Hz,1H),6.85-6.83(d,J=8.3Hz,1H),5.19(s,1H),4.32-4.27(m,1H),4.19-4.03(m,4H),3.97-3.96(d,J=4.2Hz,2H),3.72-3.66(m,5H),3.41-3.34(m,6H),2.87-2.82(ddd,J=16.5,13.1,5.6Hz,8H),2.67-2.63(t,J=7.7Hz,2H),1.44(s,3H),1.38(s,3H).13CNMR(100MHz,CDCl3):δ173.71,159.08,157.54,134.32,129.35,114.57,109.84,73.57,70.38,68.37,66.45,66.32,64.73,51.47,49.29,43.97,40.69,35.89,30.00,26.69,25.39.實施例4:本發明中加入相轉移催化劑對製備中間體的影響:按照實施例1所述步驟(1)來合成式I化合物和所述步驟(2)來合成式II化合物,其中,各組的相轉移催化劑使用情況見表1和表2,表1:製備中間體I中相轉移催化劑的用量對比組號相轉移催化劑種類及用量反應時間(h)中間體I收率1無15~1665%1a無7~840%2四正丁基碘化銨(0.10)7~882%3三乙基苄基氯化銨(0.10)7~870%4PEG400(0.10)7~870%5四正丁基溴化銨(0.10)7~885%5a四正丁基溴化銨(0.20)7~885%5b四正丁基溴化銨(0.15)7~885%5c四正丁基溴化銨(0.08)7~880%5d四正丁基溴化銨(0.05)7~870%(註:表1中相轉移催化劑的用量為其與對羥基苯丙酸的摩爾比)從表1可以看出,在式I即4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯的合成中,反應中不加入相轉移催化劑的反應時間是15h,而加入相轉移催化劑後時間縮短至7小時,且加入相轉移催化劑後式I的收率是明顯提高的,在所有相轉移催化劑中,四正丁基溴化銨的催化效果最優。在式I即4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯的合成中,當對羥基苯丙酸、氫氧化鉀、無水碳酸鉀、四正丁基溴化銨和(2,2-二甲基-1,3-二氧環戊烷-4S)甲基氯的摩爾比為1∶0.98∶1.5∶0.1∶2時,式I的收率達到最優。表2:本發明製備中間體II中相轉移催化劑的種類對比組號相轉移催化劑種類及用量反應時間(h)中間體I收率1--20~2174%1a--6~6.530%2四正丁基溴化銨(0.1)6~6.580%3四正丁基碘化銨(0.1)6~6.580%4三乙基苄基氯化銨(0.1)6~6.585%5三乙基苄基溴化銨(0.1)6~6.590%6四正丁基硫酸氫按(0.1)6~6.580%7PEG400(0.1)6~6.580%8PEG300(0.1)6~6.580%9PEG600(0.1)6~6.580%5a三乙基苄基溴化銨(0.15)6~6.584%5b三乙基苄基溴化銨(0.12)6~6.590%5c三乙基苄基溴化銨(0.05)6~6.590%5d三乙基苄基溴化銨(0.03)6~6.585%(註:表2中相轉移催化劑的用量為相轉移催化劑與中間體I的摩爾比)從表2可以看出,在式II即4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯的合成中,見表2(編號1~9及1a),不加入相轉移催化劑的反應時間是20小時,加入相轉移催化劑的反應時間是6.5小時,且加入相轉移催化劑的收率是明顯提高的,另外,在所有相轉移催化劑中,三乙基苄基溴化銨的催化效果最優。式II,即4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-(4-(2S,3-環氧丙烷)苯基)丙酯的合成中,當4S-(2,2-二甲基-1,3-二氧環戊烷-4-基)-3-(4-羥基苯基)丙酯、R-(-)-環氧氯丙烷、無水碳酸鉀和相轉移催化劑三乙基苄基溴化銨的摩爾比為1∶5∶2∶0.05時,收率達到最優。實施例5:本發明中採用的成鹽法對鹽酸蘭地洛爾的產品影響按照實施例1所述步驟(5)來合成鹽酸蘭地洛爾,其中,各組的成鹽方式見表3,表3:本發明所述鹽酸蘭地洛爾成鹽的篩選過程從表3可見,在蘭地洛爾經成鹽反應生成鹽酸蘭地洛爾的合成中,當選擇常見的成鹽方式,即在有機溶劑中成鹽,則鹽酸蘭地洛爾的收率和純度都不高,如在丙酮中通入乾燥的氯化氫氣體,鹽酸蘭地洛爾的收率和純度依次為50%,60%;此外,從表3可見,在水溶液中成鹽時,鹽酸的濃度及成鹽後體系的pH變化對鹽酸蘭地洛爾的收率和純度將有影響:鹽酸濃度過高,將生成副產物,如使用0.5~2mol/L鹽酸溶液並將pH調至4.5~6左右時,鹽酸蘭地洛爾的收率和純度為70-78%和85-95%;而使用0.1~0.3mol/L鹽酸溶液將pH調至4.5~5左右時,鹽酸蘭地洛爾的收率和純度為85%和99.5%;使用0.2mol/L鹽酸溶液將pH調至6左右時,鹽酸蘭地洛爾的收率和純度依次為80%和95%。由表3可見,在飽和NaCl水溶液和飽和NH4Cl水溶液中成鹽,用0.2mol/L鹽酸溶液將pH調至4.5~5左右時,鹽酸蘭地洛爾的收率和純度均較高。由表3可以看出:選擇飽和NH4Cl水溶液成鹽方法,靜置1小時後分液並去除瓶壁油狀物雜質,剩餘體系萃取後進一步精製,其鹽酸蘭地洛爾的收率和純度最優。以上對本發明具體實施方式的描述並不限制本發明,本領域技術人員可以根據本發明作出各種改變或變形,只要不脫離本發明的精神,均應屬於本發明所附權利要求的範圍。當前第1頁1 2 3