一種中藥組合物的指紋圖譜的測定方法與流程
2023-06-22 19:50:06 3
本發明涉及到一種中藥組合物的指紋圖譜的測定方法。
背景技術:
中藥指紋圖譜是指某些中藥材或中藥製劑經適當處理後,採用一定的分析手段,得到的能夠標示其化學特徵的色譜圖或光譜圖。中藥指紋圖譜是一種綜合的,可量化的鑑定手段,它是建立在中藥化學成分系統研究的基礎上,主要用於評價中藥材以及中藥製劑半成品質量的真實性、優良性和穩定性。「整體性」和「模糊性」為其顯著特點。以指紋圖譜作為中藥(天然藥物)提取物及其製劑的質量控制方法,已成為目前國際共識,各種符合中藥(天然藥物)特色的指紋圖譜控制技術體系正在研究和建立。美國食品藥品管理局(fda)允許草藥保健品申報資料中提供色譜指紋圖譜;世界衛生組織(who)在1996年草藥評價指導原則中也規定,如果草藥的活性成分不明確,可以提供色譜指紋圖譜以證明產品質量的一致;歐共體在草藥質量指南中亦稱,單靠測定某種有效成分考查質量的穩定性是不夠的,因為草藥及其製劑是以整體為活性物質。色譜指紋圖譜尤其是薄層色譜的鮮明的指紋圖譜是很有用的。國外指紋圖譜的應用,目的在於解決成分複雜,有效成分不明確的植物藥質量檢測和產品批次間質量差異的問題。
高效液相色譜(hplc)指紋圖譜,具有很高的分離度,可把複雜的化學成分進行分離而形成高低不同的峰組成一張色譜圖,這些色譜峰的高度和峰面積分別代表了各種不同化學成分和其含量。由此可見,中藥指紋圖譜比dna指紋圖譜更進一步的發展在於:不但有特徵的體現(各種化學成分的個數和相對位置——保留時間)可作定性鑑別使用,還體現了量的概念。峰的高度和峰面積表示了某個化學成分的含量,而各峰的峰高(或峰面積)的比值體現了各種化學成分間的相對含量;量的概念的引入、定性和定量的結合賦予中藥指紋圖譜更大的功效;中藥指紋圖譜不僅可以進行個體、某物種的「唯一性」的鑑定,還可以將其「量」的特徵和其他體系掛鈎。因此,中藥指紋圖譜不僅是一種中藥質量控制模式和技術,更可以發展成為一種採用各種指紋圖譜來進行中藥理論(複雜系統)和新藥開發的研究體系和研究模式。指紋圖譜應該具備指紋性,即:(1)專屬性強。指所制訂的指紋圖譜應該是該中藥所獨有的、能與其它中藥相區別的,其反映的化學信息是具有高度的選擇性的;(2)穩定性好。即中藥的指紋圖譜應該是從某中藥的多批次中歸納出的共性,圖譜中的共有峰或特徵峰應相對穩定;(3)重現性好。所制訂的指紋圖譜在規定條件下應能再現指紋特徵(如共有峰數目、大小、位置等),其誤差應在允許的範圍內。只有這樣,所制訂的指紋圖譜才具有實用價值,才可以有效控制藥品的質量。採用uplc指紋圖譜方法控制藥品質量具有靈敏、快速、簡便、準確等特點,通過優選色譜條件測定藥品製劑的指紋圖譜,可以很好控制藥品質量。
中藥指紋圖譜可用於中藥製劑研究、生產過程的各個階段,按應用對象來分類,可分為中藥材(原料藥材)指紋圖譜、中藥原料藥(包括飲片、配伍顆粒)指紋圖譜和中藥製劑指紋圖譜。如分得更細,還可包括用於工藝生產過程中間產物的指紋圖譜。值得注意的是建立中藥材化學指紋圖譜必須考慮諸多因素的影響。中藥品種混雜,歷史上形成的同名異物、異名同物,以及同一物種受產地環境、不同有效部位所含成分的波動大等因素,給質量控制帶來了很多困擾。因此對於某一種中藥材的指紋圖譜,需要有大量的數據積累,才能制定合理的相似性標準。中藥及其製劑均為多組分複雜體系,因此評價其質量應採用與之相適應的,能提供豐富鑑別信息的檢測方法,但現行的顯微鑑別、理化鑑別和含量測定等方法都不足以解決這一問題,建立中藥指紋圖譜將能較為全面地反映中藥及其製劑中所含化學成分的種類與數量,進而對藥品質量進行整體描述和評價。這也正好符合中醫藥整體學說。在此基礎上,如果進一步開展譜效學研究,可使中藥質量與其藥效真正結合起來,有助於闡明中藥作用機理。總之,中藥指紋圖譜的研究和建立,對於提高中藥質量,促進中藥現代化具有重要意義。
專利申請號為201210450660.0,發明名稱為:一種抗病毒中藥組合物的製備工藝及指紋圖譜的測定方法,公開了連翹、大黃、麻黃、魚腥草超聲逆流提取的提取液的指紋圖譜測定方法,標定了27個共有峰,指認了其中4個共有峰,分別為連翹酯苷a、連翹苷、松脂素-4-o-glc、大黃酸,該申請沒有公開本發明的技術內容。本發明要求保護一種中藥組合物的高效液相色譜指紋圖譜測定方法,該中藥組合物由以下藥材組成:連翹、金銀花、板藍根、苦杏仁、薄荷腦、魚腥草、大黃、廣藿香、綿馬貫眾、紅景天、麻黃、甘草、石膏,該指紋圖譜測定方法,標定了15個主要色譜峰,指認了其中9個,分別為新綠原酸,綠原酸,隱綠原酸,異連翹酯苷a,連翹酯苷a,連翹苷,槲皮苷,4,5-二-o-咖啡醯奎寧酸,連翹苷,甘草酸,而現有技術中並沒有公開該技術內容。
技術實現要素:
本發明提供一種中藥組合物的指紋圖譜測定方法。
該中藥組合物由如下重量份的原料藥製成:連翹200-300、麻黃60-100、大黃40-60、魚腥草200-300、金銀花200-300、板藍根200-300、廣藿香60-100、綿馬貫眾200-300、紅景天60-100、薄荷腦5-9、苦杏仁60-100、甘草60-100、石膏200-300,指紋圖譜測定方法如下:
供試品溶液製備:取所述中藥組合物0.1-0.5g,精密稱定,置具塞錐形瓶中,精密加入40-100%甲醇10-50ml,稱定重量,超聲處理20-40分鐘,放冷,再稱定重量,用40-100%的甲醇補足減失的重量,搖勻,濾過,取續濾液,即得;
對照品溶液的製備:分別稱取新綠原酸、綠原酸、隱綠原酸、異連翹酯苷a、連翹酯苷a、連翹苷、4,5-二-o-咖啡醯奎寧酸、槲皮苷、甘草酸銨對照品適量,製成分別含18.4µg/ml新綠原酸、27.2µg/ml綠原酸、21.2µg/ml隱綠原酸、27.2µg/ml異連翹酯苷a、21.6µg/ml連翹酯苷a、16.0µg/ml連翹苷、14.4µg/ml4,5-二-o-咖啡醯奎寧酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸銨混合對照品溶液即得;
參照物溶液的製備:取連翹酯苷a對照品適量,精密稱定,加50%甲醇製成每1ml含101.2µg的溶液,即得。
色譜條件:流動相為甲醇-乙腈-0.1%磷酸,梯度洗脫,洗脫比例如下表:
色譜柱為c18色譜柱;檢測波長239nm,流速0.1-0.5ml/min;
測定法:分別精密吸取對照品溶液與供試品溶液各1-10µl,注入超高液相色譜儀,測定,記錄色譜圖,即得。
本發明所述的中藥組合物優選的重量份原料藥為:連翹200、金銀花300、板藍根200、大黃40、廣藿香60、綿馬貫眾300、紅景天100、薄荷腦9、麻黃60、苦杏仁100、魚腥草200、甘草100、石膏200。
本發明所述的中藥組合物優選的重量份原料藥為:連翹300、金銀花200、板藍根300、大黃60、廣藿香100、綿馬貫眾200、紅景天60、薄荷腦5、麻黃100、苦杏仁60、魚腥草300、甘草60、石膏300。
本發明所述的中藥組合物優選的重量份原料藥為:連翹278、金銀花294、板藍根285、大黃55、廣藿香95、綿馬貫眾290、紅景天87、薄荷腦8.5、麻黃88、苦杏仁80、魚腥草284、甘草95、石膏277。
本發明所述的中藥組合物優選的重量份原料藥為:連翹255、金銀花255、板藍根255、大黃51、廣藿香85、綿馬貫眾255、紅景天85、薄荷腦7.5、麻黃85、苦杏仁85、魚腥草255、甘草85、石膏255。
本發明所述的中藥組合物製劑的製備方法為:
(1)按照原料藥重量比例稱取中藥材,淨選,酌情碎斷;
(2)廣藿香碎斷,加10倍量水提取揮髮油,提油時間8小時,收集揮髮油,備用;提取液過濾後,殘渣棄去,濾液備用;
(3)連翹、麻黃、魚腥草、大黃,用12倍量70%的乙醇提取3次,每次2.5小時,提取液合併過濾,回收乙醇,濾液備用;
(4)金銀花、石膏、板藍根、綿馬貫眾、甘草、紅景天,加12倍量水煎煮至沸,加入苦杏仁,煎煮2次,每次1小時,提取液合併過濾,所得濾液與步驟(2)廣藿香提油後的濾液合併,濃縮成在60℃時測定相對密度為1.10-1.15的清膏,加入乙醇,調節至醇濃度為70%,冷藏放置,過濾,回收乙醇至無醇味,得清膏備用;
(5)將步驟(4)所得清膏與步驟(3)所得醇提液合併,濃縮至在60℃時測定相對密度為1.15-1.20的清膏,乾燥,得幹膏粉,備用;
(6)將步驟(5)所得幹膏粉加入適當藥學上可接受的輔料制粒;
(7)將薄荷腦、步驟(2)所得揮髮油加入乙醇溶解,噴入步驟(6)所得顆粒,密閉,混勻,壓片或者裝膠囊、或者裝袋。
本發明所述指紋圖譜測定方法優選為:
供試品溶液製備:取所述中藥組合物0.25g,精密稱定,置具塞錐形瓶中,精密加入50%甲醇25ml,稱定重量,超聲處理(功率500w,頻率40khz)30分鐘,放冷,再稱定重量,用50%的甲醇補足減失的重量,搖勻,濾過,取續濾液,即得;
對照品溶液的製備:分別稱取新綠原酸、綠原酸、隱綠原酸、異連翹酯苷a、連翹酯苷a、連翹苷、4,5-二-o-咖啡醯奎寧酸、槲皮苷、甘草酸銨對照品適量,製成分別含18.4µg/ml新綠原酸、27.2µg/ml綠原酸、21.2µg/ml隱綠原酸、27.2µg/ml異連翹酯苷a、21.6µg/ml連翹酯苷a、16.0µg/ml連翹苷、14.4µg/ml4,5-二-o-咖啡醯奎寧酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸銨混合對照品溶液即得;
參照物溶液的製備:取連翹酯苷a對照品適量,精密稱定,加50%甲醇製成每1ml含101.2µg的溶液,即得。
色譜條件:流動相為甲醇-乙腈-0.1%磷酸,梯度洗脫,洗脫比例如下表:
色譜柱為watersacquityuplchsst3色譜柱,柱長為100mm,內徑為2.1mm,粒徑為1.8µm;檢測波長239nm,流速0.3ml/min;
測定法:分別精密吸取對照品溶液與供試品溶液各2µl,注入超高液相色譜儀,測定,記錄色譜圖,即得。
本發明所述指紋圖譜測定方法還優選為:
供試品溶液製備:取所述中藥組合物0.1g,精密稱定,置具塞錐形瓶中,精密加入甲醇50ml,稱定重量,超聲處理40分鐘,放冷,再稱定重量,用甲醇補足減失的重量,搖勻,濾過,取續濾液,即得;
對照品溶液的製備:分別稱取新綠原酸、綠原酸、隱綠原酸、異連翹酯苷a、連翹酯苷a、連翹苷、4,5-二-o-咖啡醯奎寧酸、槲皮苷、甘草酸銨對照品適量,製成分別含18.4µg/ml新綠原酸、27.2µg/ml綠原酸、21.2µg/ml隱綠原酸、27.2µg/ml異連翹酯苷a、21.6µg/ml連翹酯苷a、16.0µg/ml連翹苷、14.4µg/ml4,5-二-o-咖啡醯奎寧酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸銨混合對照品溶液即得;
參照物溶液的製備:取連翹酯苷a對照品適量,精密稱定,加50%甲醇製成每1ml含101.2µg的溶液,即得。
色譜條件:流動相為甲醇-乙腈-0.1%磷酸,梯度洗脫,洗脫比例如下表:
色譜柱為watersacquityuplchsst3色譜柱;檢測波長239nm,流速0.1ml/min;
測定法:分別精密吸取對照品溶液與供試品溶液各1µl,注入超高液相色譜儀,測定,記錄色譜圖,即得。
本發明所述指紋圖譜測定方法還優選為:
供試品溶液製備:取所述中藥組合物0.5g,精密稱定,置具塞錐形瓶中,精密加入40%甲醇10ml,稱定重量,超聲處理20分鐘,放冷,再稱定重量,用40%的甲醇補足減失的重量,搖勻,濾過,取續濾液,即得;
對照品溶液的製備:分別稱取新綠原酸、綠原酸、隱綠原酸、異連翹酯苷a、連翹酯苷a、連翹苷、4,5-二-o-咖啡醯奎寧酸、槲皮苷、甘草酸銨對照品適量,製成分別含18.4µg/ml新綠原酸、27.2µg/ml綠原酸、21.2µg/ml隱綠原酸、27.2µg/ml異連翹酯苷a、21.6µg/ml連翹酯苷a、16.0µg/ml連翹苷、14.4µg/ml4,5-二-o-咖啡醯奎寧酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸銨混合對照品溶液即得;
參照物溶液的製備:取連翹酯苷a對照品適量,精密稱定,加50%甲醇製成每1ml含101.2µg的溶液,即得。
色譜條件:流動相為甲醇-乙腈-0.1%磷酸,梯度洗脫,洗脫比例如下表:
色譜柱為watersxbridgetmshieldrp18色譜柱;檢測波長239nm,流速0.5ml/min;
測定法:分別精密吸取對照品溶液與供試品溶液各1µl,注入超高液相色譜儀,測定,記錄色譜圖,即得。
本發明所述指紋圖譜測定方法還優選為:
供試品溶液製備:取所述中藥組合物0.2g,精密稱定,置具塞錐形瓶中,精密加入90%甲醇20ml,稱定重量,超聲處理40分鐘,放冷,再稱定重量,用90%的甲醇補足減失的重量,搖勻,濾過,取續濾液,即得;
對照品溶液的製備:分別稱取新綠原酸、綠原酸、隱綠原酸、異連翹酯苷a、連翹酯苷a、連翹苷、4,5-二-o-咖啡醯奎寧酸、槲皮苷、甘草酸銨對照品適量,製成分別含18.4µg/ml新綠原酸、27.2µg/ml綠原酸、21.2µg/ml隱綠原酸、27.2µg/ml異連翹酯苷a、21.6µg/ml連翹酯苷a、16.0µg/ml連翹苷、14.4µg/ml4,5-二-o-咖啡醯奎寧酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸銨混合對照品溶液即得;
參照物溶液的製備:取連翹酯苷a對照品適量,精密稱定,加50%甲醇製成每1ml含101.2µg的溶液,即得。
色譜條件:流動相為甲醇-乙腈-0.1%磷酸,梯度洗脫,洗脫比例如下表:
色譜柱為phenomenexkinetxxb-c18;檢測波長239nm,流速0.4ml/min;
測定法:分別精密吸取對照品溶液與供試品溶液各6µl,注入超高液相色譜儀,測定,記錄色譜圖,即得。
用實施例1製備的樣品,對本發明中藥組合物的指紋圖譜測定方法,從多個方面評價其可行性,評價方法如下:
1.儀器、試劑與試藥
1.1儀器
美國watersuplch-class超高效液相色譜儀(pda檢測器,empower3.0色譜工作站),循環式多用真空泵(鄭州長城科工貿有限公司),kq250db型超聲波清洗器(崑山市超聲儀器有限公司),瑞士mettlertoledoal204型電子分析天平,瑞士mettlertoledoab135-s型電子分析天平,上海精科ja2603b電子天平。
1.2試劑
連翹苷(中國食品藥品檢定研究院,批號110821-201213,純度95.3%),連翹酯苷a(中國食品藥品檢定研究院,批號111810-201304,純度94.3%),新綠原酸(成都普瑞法科技開發有限公司,批號:13112712,純度≥98%),綠原酸(中國食品藥品檢定研究院,批號:110753-201314,純度,96.6%),隱綠原酸(上海源葉生物科技有限公司,批號:zf0226ba14,純度≥98%),異連翹酯苷a(上海源葉生物科技有限公司,批號:ya0817hb14,純度≥98%)、4,5-二-o-咖啡醯奎寧酸(中國食品藥品檢定研究院,批號:111894-201102,純度,94.1%),槲皮苷(hwianalytikgmbhpharmasolutions,批號:hwi01112,純度,91.7%),甘草酸銨(councilofeurope-edqm,批號:batch:1.1,純度≥98%),甲醇(分析純,天津市光復科技發展有限公司,批號2014年1月13日),磷酸(分析純,天津市光復科技發展有限公司,批號2014年3月26日),乙腈(色譜純,美國fisher公司,lot106246)。
1.3試藥
本發明中藥組合物(石家莊以嶺藥業股份有限公司)
2.色譜條件優化
2.1.檢測波長的選擇
在210nm~400nm考察供試品溶液不同檢測波長下的色譜圖,以239nm下的信息量較豐富,因此檢測波長最優選擇239nm,具體信息見下附圖1-4。
2.2流動相系統的選擇
本研究比較了甲醇-0.1%磷酸、乙腈-0.1%磷酸和甲醇-乙腈-0.1%磷酸三個流動相系統。結果顯示甲醇-乙腈-0.1%磷酸系統色譜峰的分離度最好,具體信息見附圖5-7,因此,確定流動相系統為甲醇-乙腈-0.1%磷酸,梯度洗脫表見表1,流速0.3ml/min。
表1流動相梯度洗脫表
3.供試品溶液和對照品溶液的製備
3.1提取溶劑的考察
分別比較了甲醇,90%甲醇,70%甲醇,50%甲醇的提取效果,結果表明50%甲醇提取所得指紋圖譜中色譜峰最多,具體信息見附圖8-11,因此選擇50%甲醇作為提取溶劑。
3.2提取方法的考察
分別比較了超聲和回流兩種提取方法,超聲和回流提取兩種方法所得的指紋圖譜色譜峰的數目和強度沒有明顯區別,具體信息見附圖12-13,由於超聲提取方法簡便,因此選擇超聲提取。
3.3超聲時間的考察
分別比較了超聲20分鐘、30分鐘、40分鐘,結果表明超聲30分鐘與40分鐘所得指紋圖譜中色譜峰信息量大,無明顯區別,具體信息見附圖14-16,因此最終確定超聲時間為30分鐘。
3.4供試品溶液製備方法的確定:取本發明中藥組合物內容物適量,研細,取0.25g,精密稱定,置具塞錐形瓶中,精密加入50%甲醇25ml,稱定重量,超聲處理(功率500w,頻率40khz)30分鐘,放冷,再稱定重量,用50%的甲醇補足減失的重量,搖勻,濾過,取續濾液,即得。
3.5對照品溶液的製備:分別稱取新綠原酸、綠原酸、隱綠原酸、異連翹酯苷a、連翹酯苷a、連翹苷、4,5-二-o-咖啡醯奎寧酸、槲皮苷、甘草酸銨對照品適量,製成分別含18.4µg/ml新綠原酸、27.2µg/ml綠原酸、21.2µg/ml隱綠原酸、27.2µg/ml異連翹酯苷a、21.6µg/ml連翹酯苷a、16.0µg/ml連翹苷、14.4µg/ml4,5-二-o-咖啡醯奎寧酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸銨混合對照品溶液即得。
3.6參照物溶液的製備:取連翹酯苷a對照品適量,精密稱定,加50%甲醇製成每1ml含101.2µg的溶液,即得。
主要色譜峰的光譜特徵和辨認
在已確定的色譜條件下,分別對對照品溶液和供試品溶液進樣,供試品溶液的色譜圖中確定16個主要色譜峰。根據色譜峰的保留時間和光譜特徵,確定指紋圖譜中2號峰為新綠原酸,3號峰為綠原酸,5號峰為隱綠原酸,8號峰為異連翹酯苷a,11號峰為連翹酯苷a,13號峰為槲皮苷,14號峰為4,5-二-o-咖啡醯奎寧酸,15號峰為連翹苷,16號峰為甘草酸,以上對照品與供試品色譜圖及光譜圖見附圖17-28。
精密度
取供試品溶液,按確定的色譜條件連續進樣6次,各主要色譜峰峰面積的rsd小於5%,表明進樣精密度良好,結果見表2。
重複性
取本發明中藥組合物內容物6份,按已確定的「供試品溶液製備方法」製備,注入液相色譜儀檢測,測定各主要色譜峰峰面積的rsd均小於5%,表明方法重複性較好,結果見表3。
穩定性
取供試品溶液,分別在0h,2h,4h,8h,12h,24h進樣,測定各主要色譜峰峰面積的rsd均小於10%,表明供試品溶液在24h內穩定,結果見表4。
指紋圖譜的建立及相似度分析
8.1指紋圖譜的建立:取10批本發明中藥組合物樣品,按3.4項下要求製備供試品溶液,在上述色譜條件下進樣分析,得到10批本發明中藥組合物的指紋圖譜,並採用國家藥典委員會開發的中藥色譜指紋圖譜相似度評價系統2004a版軟體對10批樣品的指紋圖譜進行分析,採用中位數法對各指紋圖譜色譜峰進行了多點校正和自動匹配,計算相似度並生成對照指紋圖譜,見附圖29-30,相似度結果見表5,指紋圖譜中共有色譜峰16個,用標準品指認了其中9個共有峰:新綠原酸(2號峰),綠原酸(3號峰),隱綠原酸(5號峰),異連翹酯苷a(8號峰),連翹酯苷a(11號峰),槲皮苷(13號峰),4,5-二-o-咖啡醯奎寧酸(14號峰),連翹苷(15號峰),甘草酸(16號峰)。其中以出峰穩定,峰面積較大的11號色譜峰連翹酯苷a為參照峰:
本發明建立了本發明中藥組合物的液相指紋圖譜方法,共有色譜峰16個,用標準品指認了其中9個共有峰:新綠原酸(2號峰),綠原酸(3號峰),隱綠原酸(5號峰),異連翹酯苷a(8號峰),連翹酯苷a(11號峰),槲皮苷(13號峰),4,5-二-o-咖啡醯奎寧酸(14號峰),連翹苷(15號峰),甘草酸(16號峰)。其中以出峰穩定,峰面積較大的11號色譜峰連翹酯苷a為參照峰,10批本發明中藥組合物指紋圖譜的相似度較高。由以上實驗結果可知,本方法具有良好的精密度和穩定性,以及重複性,為提高該中藥組合物的質量控制提供一種新方法。
附圖說明
圖1:檢測波長320nm下供試品溶液色譜圖
圖2:檢測波長290nm下供試品溶液色譜圖
圖3:檢測波長254nm下供試品溶液色譜圖
圖4:檢測波長230nm下供試品溶液色譜圖
圖5:甲醇-0.1%磷酸供試品溶液色譜圖
圖6:乙腈-0.1%磷酸供試品溶液色譜圖
圖7:甲醇-乙腈-0.1%磷酸供試品溶液色譜圖
圖8:50%甲醇提取供試品溶液色譜圖
圖9:70%甲醇提取供試品溶液色譜圖
圖10:90%甲醇提取供試品溶液色譜圖
圖11:甲醇提取供試品溶液色譜圖
圖12:超聲30分鐘供試品溶液色譜圖
圖13:回流1小時供試品溶液色譜圖
圖14:超聲20分鐘供試品溶液色譜圖
圖15:超聲30分鐘供試品溶液色譜圖
圖16:超聲40分鐘供試品溶液色譜圖
圖17:對照品溶液的色譜圖
圖18:連翹酯苷a參照物溶液的色譜圖
圖19:供試品溶液的色譜圖
圖20:對照品溶液與供試品溶液中新綠原酸光譜圖比較,虛線為供試品光譜圖,實線為對照品光譜圖
圖21:對照品溶液與供試品溶液中綠原酸光譜圖比較,虛線為供試品光譜圖,實線為對照品光譜圖
圖22:對照品溶液與供試品溶液中隱綠原酸光譜圖比較,虛線為供試品光譜圖,實線為對照品光譜圖
圖23:對照品溶液與供試品溶液中異連翹酯苷a光譜圖比較,虛線為供試品光譜圖,實線為對照品光譜圖
圖24:對照品溶液與供試品溶液中連翹酯苷a光譜圖比較,虛線為供試品光譜圖,實線為對照品光譜圖
圖25:對照品溶液與供試品溶液中槲皮苷光譜圖比較,虛線為供試品光譜圖,實線為對照品光譜圖
圖26:對照品溶液與供試品溶液中4,5-二-o-咖啡醯奎寧酸光譜圖比較,虛線為供試品光譜圖,實線為對照品光譜圖
圖27:對照品溶液與供試品溶液中連翹苷光譜圖比較,虛線為供試品光譜圖,實線為對照品光譜圖
圖28:對照品溶液與供試品溶液中甘草酸光譜圖比較,虛線為供試品光譜圖,實線為對照品光譜圖
圖29:10批本發明中藥組合物色譜指紋圖譜
圖30:本發明中藥組合物對照指紋圖譜
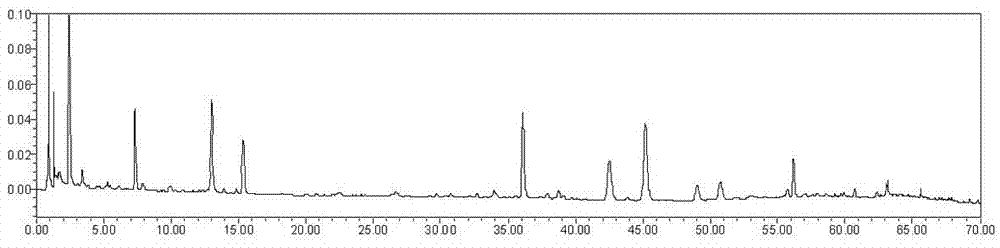
圖31:實施例1指紋圖譜,其中a圖中峰1為新綠原酸,2為綠原酸,3為隱綠原酸,4為異連翹酯苷a,5為連翹酯苷a,6為槲皮苷,7為4,5-二-o-咖啡醯奎寧酸,8為連翹苷,9為甘草酸;b圖為連翹酯苷a參照物溶液的色譜圖;c為供試品溶液指紋色譜圖
圖32:實施例2指紋圖譜,其中a圖中峰1為新綠原酸,2為綠原酸,3為隱綠原酸,4為異連翹酯苷a,5為連翹酯苷a,6為槲皮苷,7為4,5-二-o-咖啡醯奎寧酸,8為連翹苷,9為甘草酸;b圖為連翹酯苷a參照物溶液的色譜圖;c為供試品溶液指紋色譜圖
圖33:實施例3指紋圖譜,其中a圖中峰1為新綠原酸,2為綠原酸,3為隱綠原酸,4為異連翹酯苷a,5為連翹酯苷a,6為槲皮苷,7為4,5-二-o-咖啡醯奎寧酸,8為連翹苷,9為甘草酸;b圖為連翹酯苷a參照物溶液的色譜圖;c為供試品溶液指紋色譜圖
圖34:實施例4指紋圖譜,其中a圖中峰1為新綠原酸,2為綠原酸,3為隱綠原酸,4為異連翹酯苷a,5為連翹酯苷a,6為槲皮苷,7為4,5-二-o-咖啡醯奎寧酸,8為連翹苷,9為甘草酸;b圖為連翹酯苷a參照物溶液的色譜圖;c為供試品溶液指紋色譜圖。
具體實施方式
實施例1
按比例稱取:連翹200g、金銀花300g、板藍根200g、大黃40g、廣藿香60g、綿馬貫眾300g、紅景天100g、薄荷腦9g、麻黃60g、苦杏仁100g、魚腥草200g、甘草100g、石膏200g,按照以下工藝提取:
(1)按照原料藥重量比例稱取中藥材,淨選,酌情碎斷;
(2)廣藿香碎斷,加10倍量水提取揮髮油,提油時間8小時,收集揮髮油,備用;提取液過濾後,殘渣棄去,濾液備用;
(3)連翹、麻黃、魚腥草、大黃,用12倍量70%的乙醇提取3次,每次2.5小時,提取液合併過濾,回收乙醇,濾液備用;
(4)金銀花、石膏、板藍根、綿馬貫眾、甘草、紅景天,加12倍量水煎煮至沸,加入苦杏仁,煎煮2次,每次1小時,提取液合併過濾,所得濾液與步驟(2)廣藿香提油後的濾液合併,濃縮成在60℃時測定相對密度為1.10-1.15的清膏,加入乙醇,調節至醇濃度為70%,冷藏放置,過濾,回收乙醇至無醇味,得清膏備用;
(5)將步驟(4)所得清膏與步驟(3)所得醇提液合併,濃縮至在60℃時測定相對密度為1.15-1.20的清膏,乾燥,得幹膏粉,備用;
(6)將步驟(5)所得幹膏粉加入適當藥學上可接受的輔料制粒;
(7)將薄荷腦、步驟(2)所得揮髮油加入乙醇溶解,噴入步驟(6)所得顆粒,裝膠囊,即得。
指紋圖譜測定方法:
供試品溶液製備:取所述中藥組合物0.25g,精密稱定,置具塞錐形瓶中,精密加入50%甲醇25ml,稱定重量,超聲功率500w,頻率40khz,超聲30分鐘,放冷,再稱定重量,用50%的甲醇補足減失的重量,搖勻,濾過,取續濾液,即得;
對照品溶液的製備:分別稱取新綠原酸、綠原酸、隱綠原酸、異連翹酯苷a、連翹酯苷a、連翹苷、4,5-二-o-咖啡醯奎寧酸、槲皮苷、甘草酸銨對照品適量,製成分別含18.4µg/ml新綠原酸、27.2µg/ml綠原酸、21.2µg/ml隱綠原酸、27.2µg/ml異連翹酯苷a、21.6µg/ml連翹酯苷a、16.0µg/ml連翹苷、14.4µg/ml4,5-二-o-咖啡醯奎寧酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸銨混合對照品溶液即得;
參照物溶液的製備:取連翹酯苷a對照品適量,精密稱定,加50%甲醇製成每1ml含101.2µg的溶液,即得。
色譜條件:流動相為甲醇-乙腈-0.1%磷酸,梯度洗脫,洗脫比例如下表:
色譜柱為watersacquityuplchsst3色譜柱,柱長為100mm,內徑為2.1mm,粒徑為1.8µm;檢測波長239nm,流速0.3ml/min;
測定法:分別精密吸取對照品溶液與供試品溶液各2µl,注入超高液相色譜儀,測定,記錄色譜圖,即得。
結論:指紋圖譜見附圖31,結果令人滿意可以用於控制該中藥組合物的質量。
實施例2
按比例稱取:連翹300g、金銀花200g、板藍根300g、大黃60g、廣藿香100g、綿馬貫眾200g、紅景天60g、薄荷腦5g、麻黃100g、苦杏仁60g、魚腥草300g、甘草60g、石膏300g,按照以下工藝提取:
(1)按照原料藥重量比例稱取中藥材,淨選,酌情碎斷;
(2)廣藿香碎斷,加10倍量水提取揮髮油,提油時間8小時,收集揮髮油,備用;提取液過濾後,殘渣棄去,濾液備用;
(3)連翹、麻黃、魚腥草、大黃,用12倍量70%的乙醇提取3次,每次2.5小時,提取液合併過濾,回收乙醇,濾液備用;
(4)金銀花、石膏、板藍根、綿馬貫眾、甘草、紅景天,加12倍量水煎煮至沸,加入苦杏仁,煎煮2次,每次1小時,提取液合併過濾,所得濾液與步驟(2)廣藿香提油後的濾液合併,濃縮成在60℃時測定相對密度為1.10-1.15的清膏,加入乙醇,調節至醇濃度為70%,冷藏放置,過濾,回收乙醇至無醇味,得清膏備用;
(5)將步驟(4)所得清膏與步驟(3)所得醇提液合併,濃縮至在60℃時測定相對密度為1.15-1.20的清膏,乾燥,得幹膏粉,備用;
(6)將步驟(5)所得幹膏粉加入適當藥學上可接受的輔料制粒;
(7)將薄荷腦、步驟(2)所得揮髮油加入乙醇溶解,噴入步驟(6)所得顆粒,壓片,即得。
指紋圖譜測定方法:
供試品溶液製備:取所述中藥組合物0.1g,精密稱定,置具塞錐形瓶中,精密加入100%甲醇50ml,稱定重量,超聲處理40分鐘,放冷,再稱定重量,用100%的甲醇補足減失的重量,搖勻,濾過,取續濾液,即得;
對照品溶液的製備:分別稱取新綠原酸、綠原酸、隱綠原酸、異連翹酯苷a、連翹酯苷a、連翹苷、4,5-二-o-咖啡醯奎寧酸、槲皮苷、甘草酸銨對照品適量,製成分別含18.4µg/ml新綠原酸、27.2µg/ml綠原酸、21.2µg/ml隱綠原酸、27.2µg/ml異連翹酯苷a、21.6µg/ml連翹酯苷a、16.0µg/ml連翹苷、14.4µg/ml4,5-二-o-咖啡醯奎寧酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸銨混合對照品溶液即得;
參照物溶液的製備:取連翹酯苷a對照品適量,精密稱定,加50%甲醇製成每1ml含101.2µg的溶液,即得。
色譜條件:流動相為甲醇-乙腈-0.1%磷酸,梯度洗脫,洗脫比例如下表:
色譜柱為watersacquityuplchsst3(柱長為100mm,內徑為2.1mm,粒徑為1.8µm)色譜柱;檢測波長239nm,流速0.1ml/min;
測定法:分別精密吸取對照品溶液與供試品溶液各1µl,注入超高液相色譜儀,測定,記錄色譜圖,即得。
結論:指紋圖譜見附圖32,結果令人滿意可以用於控制該中藥組合物的質量。
實施例3
原料藥配方為:連翹278g、金銀花294g、板藍根285g、大黃55g、廣藿香95g、綿馬貫眾290g、紅景天87g、薄荷腦8.5g、麻黃88g、苦杏仁80g、魚腥草284g、甘草95g、石膏277g,按照以下工藝提取:
(1)按照原料藥重量比例稱取中藥材,淨選,酌情碎斷;
(2)廣藿香碎斷,加10倍量水提取揮髮油,提油時間8小時,收集揮髮油,備用;提取液過濾後,殘渣棄去,濾液備用;
(3)連翹、麻黃、魚腥草、大黃,用12倍量70%的乙醇提取3次,每次2.5小時,提取液合併過濾,回收乙醇,濾液備用;
(4)金銀花、石膏、板藍根、綿馬貫眾、甘草、紅景天,加12倍量水煎煮至沸,加入苦杏仁,煎煮2次,每次1小時,提取液合併過濾,所得濾液與步驟(2)廣藿香提油後的濾液合併,濃縮成在60℃時測定相對密度為1.10-1.15的清膏,加入乙醇,調節至醇濃度為70%,冷藏放置,過濾,回收乙醇至無醇味,得清膏備用;
(5)將步驟(4)所得清膏與步驟(3)所得醇提液合併,濃縮至在60℃時測定相對密度為1.15-1.20的清膏,乾燥,得幹膏粉,備用;
(6)將步驟(5)所得幹膏粉加入適當藥學上可接受的輔料制粒;
(7)將薄荷腦、步驟(2)所得揮髮油加入乙醇溶解,噴入步驟(6)所得顆粒,裝袋,即得。
指紋圖譜測定方法:
供試品溶液製備:取所述中藥組合物0.5g,精密稱定,置具塞錐形瓶中,精密加入40%甲醇10ml,稱定重量,超聲處理20分鐘,放冷,再稱定重量,用40%的甲醇補足減失的重量,搖勻,濾過,取續濾液,即得;
對照品溶液的製備:分別稱取新綠原酸、綠原酸、隱綠原酸、異連翹酯苷a、連翹酯苷a、連翹苷、4,5-二-o-咖啡醯奎寧酸、槲皮苷、甘草酸銨對照品適量,製成分別含18.4µg/ml新綠原酸、27.2µg/ml綠原酸、21.2µg/ml隱綠原酸、27.2µg/ml異連翹酯苷a、21.6µg/ml連翹酯苷a、16.0µg/ml連翹苷、14.4µg/ml4,5-二-o-咖啡醯奎寧酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸銨混合對照品溶液即得;
參照物溶液的製備:取連翹酯苷a對照品適量,精密稱定,加50%甲醇製成每1ml含101.2µg的溶液,即得。
色譜條件:流動相為甲醇-乙腈-0.1%磷酸,梯度洗脫,洗脫比例如下表:
色譜柱為watersxbridgetmshieldrp18(5µm,4.6mm×250mm)色譜柱;檢測波長239nm,流速0.5ml/min;
測定法:分別精密吸取對照品溶液與供試品溶液各1µl,注入超高液相色譜儀,測定,記錄色譜圖,即得。
結果:指紋圖譜見附圖33,結果令人滿意可以用於控制該中藥組合物的質量。
實施例4
原料藥配方為:按比例稱取:連翹255g、金銀花255g、板藍根255g、大黃51g、廣藿香85g、綿馬貫眾255g、紅景天85g、薄荷腦7.5g、麻黃85g、苦杏仁85g、魚腥草255g、甘草85g、石膏255g,按照以下工藝提取:
(1)按照原料藥重量比例稱取中藥材,淨選,酌情碎斷;
(2)廣藿香碎斷,加10倍量水提取揮髮油,提油時間8小時,收集揮髮油,備用;提取液過濾後,殘渣棄去,濾液備用;
(3)連翹、麻黃、魚腥草、大黃,用12倍量70%的乙醇提取3次,每次2.5小時,提取液合併過濾,回收乙醇,濾液備用;
(4)金銀花、石膏、板藍根、綿馬貫眾、甘草、紅景天,加12倍量水煎煮至沸,加入苦杏仁,煎煮2次,每次1小時,提取液合併過濾,所得濾液與步驟(2)廣藿香提油後的濾液合併,濃縮成在60℃時測定相對密度為1.10-1.15的清膏,加入乙醇,調節至醇濃度為70%,冷藏放置,過濾,回收乙醇至無醇味,得清膏備用;
(5)將步驟(4)所得清膏與步驟(3)所得醇提液合併,濃縮至在60℃時測定相對密度為1.15-1.20的清膏,乾燥,得幹膏粉,備用;
(6)將步驟(5)所得幹膏粉加入適當藥學上可接受的輔料制粒;
(7)將薄荷腦、步驟(2)所得揮髮油加入乙醇溶解,噴入步驟(6)所得顆粒,裝膠囊,即得。
指紋圖譜測定方法:
供試品溶液製備:取所述中藥組合物0.2g,精密稱定,置具塞錐形瓶中,精密加入90%甲醇20ml,稱定重量,超聲處理40分鐘,放冷,再稱定重量,用90%的甲醇補足減失的重量,搖勻,濾過,取續濾液,即得;
對照品溶液的製備:分別稱取新綠原酸、綠原酸、隱綠原酸、異連翹酯苷a、連翹酯苷a、連翹苷、4,5-二-o-咖啡醯奎寧酸、槲皮苷、甘草酸銨對照品適量,製成分別含18.4µg/ml新綠原酸、27.2µg/ml綠原酸、21.2µg/ml隱綠原酸、27.2µg/ml異連翹酯苷a、21.6µg/ml連翹酯苷a、16.0µg/ml連翹苷、14.4µg/ml4,5-二-o-咖啡醯奎寧酸、9.0µg/ml槲皮苷、24.4µg/ml甘草酸銨混合對照品溶液即得;
參照物溶液的製備:取連翹酯苷a對照品適量,精密稱定,加50%甲醇製成每1ml含101.2µg的溶液,即得。
色譜條件:流動相為甲醇-乙腈-0.1%磷酸,梯度洗脫,洗脫比例如下表:
色譜柱為phenomenexkinetxxb-c18色譜柱;檢測波長239nm,流速0.4ml/min;
測定法:分別精密吸取對照品溶液與供試品溶液各6µl,注入超高液相色譜儀,測定,記錄色譜圖,即得。
結論:指紋圖譜見附圖34,結果令人滿意可以用於控制該中藥組合物的質量。