朝藥腎炎草指紋圖譜的建立方法及其指紋圖譜與流程
2023-06-10 19:19:31 4
本發明涉及民族藥材質量控制領域,具體涉及朝藥腎炎草指紋圖譜的建立方法及其指紋圖譜。
背景技術:
我國的民族醫藥之一的朝醫藥是我國歷史文化遺產的瑰寶,是在延邊這一特殊的地理環境下,朝鮮族醫學家在掌握古朝鮮東醫學理論和技術基礎上,在長期的與疾病作鬥爭的過程中形成和發展的既區別於朝鮮東醫學又不同於中醫學的較完整的醫學體系。它豐富了中華民族的醫藥學寶庫,為民族藥新藥研究提供了一個巨大的資源庫。朝藥的研究與開發在我國醫藥產業發展中具有明顯的區域特色和代表性。
民族藥的質量標準是民族藥產品核心競爭力的基本要素,是發展民族藥產業的重要技術保證。在國家中醫藥管理局等大力支持下,近年來朝藥的理論、物質基礎、臨床應用等方面取得了很大的成果,但在品種的整理和質量控制上仍然存在嚴重的標準缺失,制約了朝藥的發展。制訂全面、系統、科學合理的朝藥質量標準,不僅推進朝藥生產的規範化、標準化、現代化產生積極而深遠的影響,對控制朝藥質量,保證臨床用藥的安全有效有著極其重要的意義。
朝藥腎炎草是石竹科植物光萼女婁菜(melandriumfirmumrohrb.)及其變型疏毛女婁菜(melandriumpubescensmakino.)的乾燥地上部分,廣泛分布於長白山地區,植物資源豐富,也有大量栽培。朝藥腎炎草為朝醫學中少陽人要藥,在《東醫寶鑑》、《朝醫學》、《朝鮮藥典》、《實用東藥學》、《中國民族藥志》、《朝藥志》等民族藥古書籍文獻中均有記載,並且具有清熱解毒,活血利尿、止血調經,祛風止痛等功效,用於急、慢性腎炎、尿路感染,月經不調,關節炎,子宮出血等疾病。
目前為止,國內外對該民族藥的質量標準研究尚未見任何報導,故對其進行高效液相(hplc)指紋圖譜研究,彌補腎炎草質量標準項下的空白,為全面建立腎炎草的質量標準提供科學依據。
技術實現要素:
本發明設計開發了朝藥腎炎草指紋圖譜的建立方法,本發明的發明目的是通過朝藥腎炎草高效液相指紋圖譜的建立方法,能夠為朝藥腎炎草的鑑別和質量控制提供可靠依據。
本發明設計開發了朝藥腎炎草指紋圖譜的建立方法得到的指紋圖譜,本發明的發明目的是通過得到的朝藥腎炎草高效液相指紋圖譜,能夠為朝藥腎炎草的鑑別和質量控制提供可靠依據。
本發明提供的技術方案為:
朝藥腎炎草指紋圖譜的建立方法,包括如下步驟:
取夏佛塔苷、葒草苷、牡荊素對照品,加甲醇溶解,定容,作為對照品溶液;
取腎炎草藥材,用乙醇溶液提取,定容,作為供試品溶液;
經高效液相分離檢測,以體積分數0.4%乙酸溶液為a相,體積分數100%乙腈溶液為b相,組成流動相,採用梯度洗脫,以相對保留時間和相對峰面積標定指紋特徵,從而得到所述指紋圖譜。
優選的是,所述高效液相分離檢測條件為:採用色譜柱以十八烷基矽烷鍵合矽膠為固定相,柱溫為50℃,檢測波長為253nm,流速為1ml/min,分析時間為70min,採用進樣量為10μl注入高效液相色譜儀。
優選的是,所述梯度洗脫的洗脫程序如下:
0分鐘時,流動相a為90%的乙酸溶液,流動相b為10%的乙腈溶液;
35分鐘時,流動相a為75%的乙酸溶液,流動相b為25%的乙腈溶液;
50分鐘時,流動相a為70%的乙酸溶液,流動相b為30%的乙腈溶液;
60分鐘時,流動相a為60%的乙酸溶液,流動相b為40%的乙腈溶液;
70分鐘時,流動相a為10%的甲醇溶液,流動相b為90%的乙腈溶液;
柱溫為50℃;流速:1ml/min;進樣量:10μl。
優選的是,朝藥腎炎草供試品溶液的製備工藝包括:取腎炎草藥材粉末1.0g,精密稱定,置25ml的容量瓶中加入70%乙醇溶液,稱定初始重量,室溫下超聲lh,冷卻至室溫,用乙醇溶液補足減失重量,搖勻,用0.45μm微孔濾膜過濾,即得供試品。
優選的是,對照品溶液的製備工藝包括:精密稱定夏佛塔苷、葒草苷、牡荊素對照品,分別稱取5mg,5mg,2mg依次置於10ml、25ml、10ml容量瓶中,加甲醇稀釋至刻度,搖勻,定容,製成濃度分別為500ppm,200ppm,200ppm的溶液,經0.45μm微孔濾膜濾過,即得對照品溶液備用。
優選的是,所述指紋圖譜共有21個共有指紋峰,相對保留時間分別為:
1號峰:0.281;2號峰:0.368;3號峰:0.444;4號峰:0.603;5號峰:0.824;6號峰為參照峰:1.000;7號峰:1.053;8號峰:1.079;9號峰:1.150;10號峰:1.185;11號峰:1.206;12號峰:1.225;13號峰:1.271;14號峰:1.332;15號峰:1.362;16號峰:1.380;17號峰:1.413;18號峰:1.498;19號峰:1.559;20號峰:1.741;21號峰:1.768。
優選的是,所述指紋圖譜共有21個共有指紋峰,相對峰面積分別為:
1號峰:0.662;2號峰:0.744;3號峰:0.303;4號峰:0.092;5號峰:0.242;6號峰為參照峰:1.000;7號峰:0.152;8號峰:0.061;9號峰:0.398;10號峰:1.015;11號峰:0.136;12號峰:0.370;13號峰:1.671;14號峰:0.547;15號峰:0.189;16號峰:0.318;17號峰:0.133;18號峰:0.177;19號峰:0.107;20號峰:0.159;21號峰:1.907。
朝藥腎炎草指紋圖譜的建立方法得到的指紋圖譜,將朝藥腎炎草製備成乙醇溶液後,經過高效液相分離檢測,獲得所述朝藥腎炎草的指紋圖譜。
優選的是,所述高效液相分離檢測條件為:採用色譜柱以十八烷基矽烷鍵合矽膠為固定相,以體積分數0.4%乙酸溶液為a相,體積分數100%乙腈溶液為b相,組成流動相,採用梯度洗脫,柱溫為50℃,檢測波長為253nm,流速為1ml/min,分析時間為70min,採用進樣量為10μl注入高效液相色譜儀;以及
所述梯度洗脫的洗脫程序如下:
0分鐘時,流動相a為90%的乙酸溶液,流動相b為10%的乙腈溶液;
35分鐘時,流動相a為75%的乙酸溶液,流動相b為25%的乙腈溶液;
50分鐘時,流動相a為70%的乙酸溶液,流動相b為30%的乙腈溶液;
60分鐘時,流動相a為60%的乙酸溶液,流動相b為40%的乙腈溶液;
70分鐘時,流動相a為10%的甲醇溶液,流動相b為90%的乙腈溶液。
優選的是,所述指紋圖譜共有21個共有指紋峰;
所述共有指紋峰的相對保留時間分別為:1號峰:0.281;2號峰:0.368;3號峰:0.444;4號峰:0.603;5號峰:0.824;6號峰為參照峰:1.000;7號峰:1.053;8號峰:1.079;9號峰:1.150;10號峰:1.185;11號峰:1.206;12號峰:1.225;13號峰:1.271;14號峰:1.332;15號峰:1.362;16號峰:1.380;17號峰:1.413;18號峰:1.498;19號峰:1.559;20號峰:1.741;21號峰:1.768;
所述共有指紋峰的相對峰面積分別為:1號峰:0.662;2號峰:0.744;3號峰:0.303;4號峰:0.092;5號峰:0.242;6號峰為參照峰:1.000;7號峰:0.152;8號峰:0.061;9號峰:0.398;10號峰:1.015;11號峰:0.136;12號峰:0.370;13號峰:1.671;14號峰:0.547;15號峰:0.189;16號峰:0.318;17號峰:0.133;18號峰:0.177;19號峰:0.107;20號峰:0.159;21號峰:1.907。
本發明與現有技術相比較所具有的有益效果:本發明依據標準藥材和延邊不同產地9批供試品檢測結果,選擇了其保留時間和峰面積穩定的21個共有峰標定為腎炎草化學成分的指紋圖譜,且共有峰峰面積大於總峰面積的90%,非共有峰峰面積小於總峰面積的10%,表明具有良好的系統性、特徵性和重複性。經過相似度評價,所選擇的延邊不同縣市腎炎草指紋圖譜相似度基本都大於0.90,具有良好的相關性,完成了延邊地產腎炎草藥材指紋圖譜的建立。該方法可靠,簡便,可作為腎炎草的鑑定與質量控制的有效方法。
附圖說明
圖1為dad檢測器不同波長下腎炎草的高效液相色譜圖。
圖2為對照品供試液的高效液相色譜圖。
圖3為朝藥腎炎草標準藥材溶液的高效液相色譜圖。
圖4為10批朝藥腎炎草的高效液相指紋圖譜;其中,r:共有圖譜;s1:標準藥材;s2:龍井市;s3:汪清縣;s4:安圖縣;s5:琿春市;s6:圖們市;s7:敦化市;s8:延吉市1;s9:延吉市2;s10:和龍縣。
圖5為朝藥腎炎草共有峰指紋圖譜。
圖6為朝藥腎炎草精密度實驗高效液相色圖譜。
圖7為朝藥腎炎草重複性實驗高效液相色圖譜。
圖8為朝藥腎炎草穩定性實驗高效液相色圖譜。
具體實施方式
下面結合附圖對本發明做進一步的詳細說明,以令本領域技術人員參照說明書文字能夠據以實施。
如圖1~8所示,本發明提供朝藥腎炎草指紋圖譜的建立方法,包括如下步驟:
取夏佛塔苷、葒草苷、牡荊素對照品,加甲醇溶解,定容,作為對照品溶液;
取腎炎草藥材,用乙醇溶液提取,定容,作為供試品溶液;
經高效液相分離檢測,以體積分數0.4%乙酸溶液為a相,體積分數100%乙腈溶液為b相,組成流動相,採用梯度洗脫,以相對保留時間和相對峰面積標定指紋特徵,從而得到所述指紋圖譜。
在另一種實施例中,高效液相分離檢測條件為:採用色譜柱以十八烷基矽烷鍵合矽膠為固定相,柱溫為50℃,檢測波長為253nm,流速為1ml/min,分析時間為70min,採用進樣量為10μl注入高效液相色譜儀。
在另一種實施例中,梯度洗脫的洗脫程序如下:
0分鐘時,流動相a為90%的乙酸溶液,流動相b為10%的乙腈溶液;
35分鐘時,流動相a為75%的乙酸溶液,流動相b為25%的乙腈溶液;
50分鐘時,流動相a為70%的乙酸溶液,流動相b為30%的乙腈溶液;
60分鐘時,流動相a為60%的乙酸溶液,流動相b為40%的乙腈溶液;
70分鐘時,流動相a為10%的甲醇溶液,流動相b為90%的乙腈溶液;
所述柱溫為50℃;所述流速:1ml/min;所述進樣量:10μl。
在另一種實施例中,朝藥腎炎草供試品溶液的製備工藝包括:取腎炎草標準藥材粉末1.0g,精密稱定,置25ml的容量瓶中加入70%乙醇溶液,稱定初始重量,室溫下超聲lh,冷卻至室溫,用乙醇溶液補足減失重量,搖勻,用0.45μm微孔濾膜過濾,即得供試品。
在另一種實施例中,對照品溶液的製備工藝包括:精密稱定夏佛塔苷、葒草苷、牡荊素對照品,分別稱取5mg,5mg,2mg依次置於10ml、25ml、10ml容量瓶中,加甲醇稀釋至刻度,搖勻,定容,製成濃度分別為500ppm,200ppm,200ppm的溶液,經0.45μm微孔濾膜濾過,即得對照品溶液備用。
在另一種實施例中,指紋圖譜共有21個共有指紋峰,相對保留時間分別為:1號峰:0.281;2號峰:0.368;3號峰:0.444;4號峰:0.603;5號峰:0.824;6號峰為參照峰:1.000;7號峰:1.053;8號峰:1.079;9號峰:1.150;10號峰:1.185;11號峰:1.206;12號峰:1.225;13號峰:1.271;14號峰:1.332;15號峰:1.362;16號峰:1.380;17號峰:1.413;18號峰:1.498;19號峰:1.559;20號峰:1.741;21號峰:1.768;相對峰面積分別為:1號峰:0.662;2號峰:0.744;3號峰:0.303;4號峰:0.092;5號峰:0.242;6號峰為參照峰:1.000;7號峰:0.152;8號峰:0.061;9號峰:0.398;10號峰:1.015;11號峰:0.136;12號峰:0.370;13號峰:1.671;14號峰:0.547;15號峰:0.189;16號峰:0.318;17號峰:0.133;18號峰:0.177;19號峰:0.107;20號峰:0.159;21號峰:1.907。
本發明還提供了朝藥腎炎草指紋圖譜的建立方法得到的指紋圖譜,將朝藥腎炎草製備成乙醇溶液後,經過高效液相分離檢測,獲得所述朝藥腎炎草的指紋圖譜。
在另一種實施例中,所述高效液相分離檢測條件為:採用色譜柱以十八烷基矽烷鍵合矽膠為固定相,以體積分數0.4%乙酸溶液為a相,體積分數100%乙腈溶液為b相,組成流動相,採用梯度洗脫,柱溫為50℃,檢測波長為253nm,流速為1ml/min,分析時間為70min,採用進樣量為10μl注入高效液相色譜儀;梯度洗脫的洗脫程序如下:
0分鐘時,流動相a為90%的乙酸溶液,流動相b為10%的乙腈溶液;
35分鐘時,流動相a為75%的乙酸溶液,流動相b為25%的乙腈溶液;
50分鐘時,流動相a為70%的乙酸溶液,流動相b為30%的乙腈溶液;
60分鐘時,流動相a為60%的乙酸溶液,流動相b為40%的乙腈溶液;
70分鐘時,流動相a為10%的甲醇溶液,流動相b為90%的乙腈溶液。
在另一種實施例中,指紋圖譜共有21個共有指紋峰;
所述共有指紋峰的相對保留時間分別為:1號峰:0.281;2號峰:0.368;3號峰:0.444;4號峰:0.603;5號峰:0.824;6號峰為參照峰:1.000;7號峰:1.053;8號峰:1.079;9號峰:1.150;10號峰:1.185;11號峰:1.206;12號峰:1.225;13號峰:1.271;14號峰:1.332;15號峰:1.362;16號峰:1.380;17號峰:1.413;18號峰:1.498;19號峰:1.559;20號峰:1.741;21號峰:1.768;共有指紋峰的相對峰面積分別為:1號峰:0.662;2號峰:0.744;3號峰:0.303;4號峰:0.092;5號峰:0.242;6號峰為參照峰:1.000;7號峰:0.152;8號峰:0.061;9號峰:0.398;10號峰:1.015;11號峰:0.136;12號峰:0.370;13號峰:1.671;14號峰:0.547;15號峰:0.189;16號峰:0.318;17號峰:0.133;18號峰:0.177;19號峰:0.107;20號峰:0.159;21號峰:1.907。
1、材料
1.1供試品藥材
標準藥材(s1):由延邊民族醫藥研究所提供;s2:龍井市;s3:汪清縣;s4:安圖縣;s5:琿春市;s6:圖們市;s7:敦化市;s8:延吉市1;s9:延吉市2;s10:和龍縣。
1.2對照品
夏佛塔苷對照品(51938-32-0,上海融禾醫藥科技有限公司),葒草苷對照品(28608-75-5,上海融禾醫藥科技有限公司),牡荊素對照品(3681-93-4,上海融禾醫藥科技有限公司)。
1.3試劑
乙腈(色譜級,fisherscientific,美國),甲醇(色譜級,fisherscientific,美國),乙醇(分析純,遼寧泉瑞試劑有限公司),甲醇(分析純,遼寧泉瑞試劑有限公司),甲酸(分析純,天津市科密歐化學試劑有限公司),三氟乙酸(色譜級,上海阿拉丁生化科技股份有限公司),乙酸(冰乙酸,分析純,公主嶺市化學試劑廠),蒸餾水、超純水(延邊大學自製)。
1.4儀器
cp214型電子分析天平(奧豪斯儀器(常州)有限公司),抽濾真空裝置(上海昕滬實驗設備有限公司),移液槍(100μl,河南兄弟儀器設備有限公司),移液槍(200μl,法國),hitachi高效液相色譜儀(日本日立公司),primaide-1410紫外檢測器(日本日立公司),primaide-5430二級陣列管(dad)檢測器(日本日立公司),工作站:primaide色譜工作站(日本日立公司)。
2、方法與結果
2.1色譜條件
2.1.1檢測波長的選擇
如圖1所示,為了在指紋圖譜中獲得最豐富的腎炎草化學成分信息,利用二極體陣列檢測器(dad)檢測器對200~400nm範圍內進行全波段掃描。結果顯示在253nm下指紋圖譜的色譜峰數目最多,代表的化學成分信息量最大,峰的分離度也較好,所以最終確定253nm為檢測波長,如圖1所示。
2.1.2流動相的選擇
分別以乙腈-水和甲醇-水系統為流動相進行梯度洗脫,結果顯示乙腈-水溶劑系統洗脫所得的譜圖效果更好,但分離度仍然不夠理想,為了改善其峰形,嘗試了在水相中添加甲酸、乙酸和三氟乙酸溶液來改善分離度。結果顯示0.4%乙酸溶液作為流動相中的水相時峰形最好,所以選取乙腈-0.4%乙酸溶液作為流動相洗脫時間程序如表1所示。
表1.流動相洗脫系統
2.1.3實驗條件的確定
色譜柱為sunfirec18(5μm,4.6×250mm);流動相溶劑系統中b為乙腈溶液(acn),a為0.4%乙酸溶液(hac)(a+b=100%);流速1ml/min;柱溫50℃;檢測波長253nm;進樣量10μl;色譜記錄時間為70min,表2所示。
表2.hplc的色譜條件
2.2樣品溶液製備
2.2.1對照品溶液的製備
精密稱定夏佛塔苷、葒草苷、牡荊素對照品,分別為5mg,5mg,2mg依次置於10ml、25ml、10ml容量瓶中,加甲醇稀釋至刻度,搖勻,定容,製成濃度分別為500ppm,200ppm,200ppm的溶液,經0.45μm微孔濾膜濾過,備用。
2.2.2供試品溶液的製備
取腎炎草藥材和9批供試品藥材粉末約1.0g,精密稱定,置25ml的容量瓶中加入70%乙醇溶液,稱定初始重量,室溫下超聲l小時,冷卻至室溫,用乙醇溶液補足減失重量,搖勻,用0.45μm微孔濾膜過濾,即得供試品。
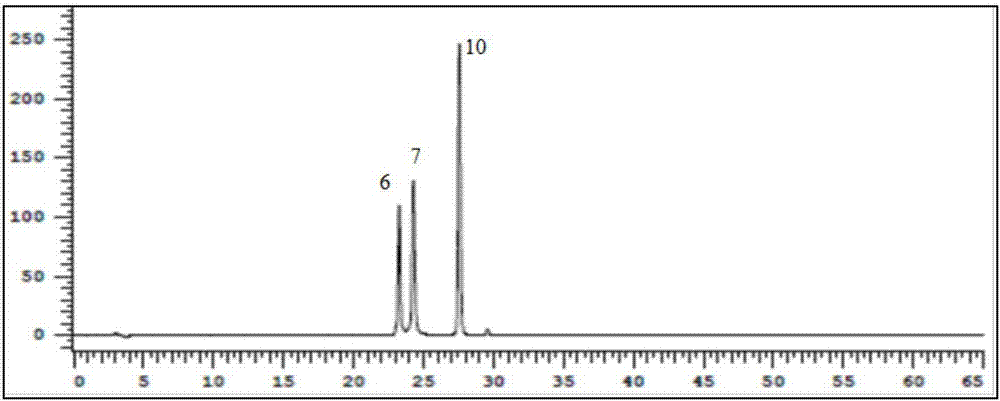
2.3朝藥腎炎草指紋圖譜建立
如圖2、圖3所示,依以上色譜條件,對1~10批供試品進樣分析,確定了分離度大於1.0的共有峰21個,對照品混合液的圖譜通過保留時間定性可知,標準藥材中峰6,7,10分別為夏佛塔苷,葒草苷和牡荊素的色譜峰;其中,夏佛塔苷的色譜峰(峰6)含量較高,且保留時間穩定,將其作為參照峰;10批腎炎草21個共有峰的相對保留時間及相對峰面積如表3,表4所示;其中,相對保留時間=各組分峰的保留時間/參照峰的保留時間,相對保留峰面積=各組分峰的峰面積/參照峰的峰面積;
表310批腎炎草指紋圖譜中共有指紋峰的相對保留時間
表410批腎炎草指紋圖譜中共有指紋峰的相對峰面積
由圖表可知,延邊地區不同產地的腎炎草藥材hplc指紋圖譜共有峰的相對保留時間的rsd為0.04-0.2%,小於3%,說明了重現性很好;但是相對峰面積值存在較為顯著的差異(rsd為17-81%),這說明了10批不同產地藥材因為其生產環境的不同導致了其化學成分含量的差別。
2.4相似度的計算
分別按「2.2」項下方法製備供試品溶液,進樣分析,利用《中國藥典》「中藥色譜指紋圖譜相似度評價系統2004a版」,將1~10批腎炎草供試品圖譜數據導入,經多點校正和數據匹配,以中位數生成對照指紋圖譜,建立共有模式的指紋圖譜(圖5),根據匹配的數據,確定了21個共有峰,對共有峰的相對保留時間進行了分析,其21個共有峰的相對保留時間的rsd在0.1%~1%之間,各產地藥材的共有峰保留時間基本一致(圖4)。
2.5不同產地之間的相似度比較
根據以上提到的色譜條件,分別將延邊不同產地的腎炎草供試品進行分析,記錄色譜圖,利用《中國藥典》「中藥色譜指紋圖譜相似度評價系統2004b版」,將1~10批腎炎草藥材圖譜生成的對照譜圖導入,計算延邊不同產地樣品與共有模式間的的相似度比較,結果如表5所示。
表5.延邊不同產地腎炎草藥材的相似度
2.5方法學驗證
2.5.1精密度實驗
精密吸取同一份供試品溶液1.0ml,按以上色譜條件連續進樣5次,經「中藥色譜指紋圖譜相似度評價系統a版」對各主要色譜峰的峰面積和保留時間考察計算,結果顯示,其主要色譜峰的峰面積以及保留時間相對標準偏差均小於3%,表明儀器精密度良好,如圖6、表6所示。
表6方法精密度測試
2.5.2重複性實驗
精密吸取同一份供試品溶液5份,按以上色譜條件分別進樣,經「中藥色譜指紋圖譜相似度評價系統a版」對各主要色譜峰的峰面積和保留時間考察計算,結果顯示其主要色譜峰的峰面積以及保留時間相對標準偏差均小於3%,說明方法重複性良好,如圖7、表7所示。
表7方法重複性測試
2.5.3穩定性實驗
精密吸取同一份供試品溶液,按以上色譜條件,分別在1、4、8、12、24h進樣,經「中藥色譜指紋圖譜相似度評價系統a版」對各主要色譜峰的峰面積和保留時間考察計算,結果顯示其主要色譜峰的峰面積以及保留時間相對標準偏差均小於3%,表明供試品溶液在24h小時內基本穩定,如圖8、表8所示。
表8樣品穩定性測試
儘管本發明的實施方案已公開如上,但其並不僅僅限於說明書和實施方式中所列運用,它完全可以被適用於各種適合本發明的領域,對於熟悉本領域的人員而言,可容易地實現另外的修改,因此在不背離權利要求及等同範圍所限定的一般概念下,本發明並不限於特定的細節和這裡示出與描述的圖例。