包含微晶纖維素的可直接壓制的組合物的製作方法
2023-11-06 14:37:04 3
背景技術:
聚乙烯醇(PVA)是特別關於其聚合度及其粘度能夠以各種品質獲得的合成聚合物。
聚乙烯醇通過聚乙酸乙烯酯的鹼性水解獲得。聚乙酸乙烯酯又通過乙酸乙烯酯的自由基聚合獲得。通過不同鏈長和不同水解度的所使用的聚乙酸乙烯酯可以獲得具有各種物理性能的聚乙烯醇(PVA)。
聚乙烯醇特別在多個應用領域(例如油漆、紙、織物、化妝品以及製藥(包括給藥體系)等)中用作成膜劑、粘附凝膠和粘度調節劑。
在製藥工業中特別感興趣的是PVA在製藥製劑(例如眼科製劑)中作為包衣片劑的成膜劑、作為顆粒中的粘合劑或作為膏藥中的塗層組分以及在給藥體系中的用途。非常特別感興趣的是各種類型的PVA在具有延長的活性成分釋放的固體口服製藥劑型的製劑(例如所謂的「緩釋片劑」)中的用途。在所述片劑中,活性成分在PVA基質中以微細分布的形式存在。
在口服之後,在所述含聚合物的製藥製劑中以如下方式實現延緩的活性成分釋放:片劑在例如口或胃腸道中的液體的存在下不直接溶解而是溶脹並且形成凝膠,僅通過擴散和凝膠基質在胃腸道中的緩慢侵蝕從所述凝膠中逐步釋放活性成分。緩釋片劑的延緩的活性成分釋放又造成近似恆定的活性成分血液水平,因此造成更好的治療效果。
這意味著通過所述蓋倫改性片劑,能夠以受控方式在更長的時間內從劑型向體內釋放活性成分,從而在更長的時間(數小時)內保持有治療效果的藥物血液水平。
不同於在服用之後立即釋放活性成分的片劑,所述緩釋製劑的兩個主要優點在於:一方面避免了不希望的可能具有毒性的API(API:藥用活性成分)血液水平/血漿水平,並且片劑的服用頻率降低(例如僅1次/天,而不是3次/天),因此改進所謂的「患者依從性」,同時改進藥物處理的治療結果。
然而,根據各種藥典(歐洲藥典Ph.Eur;美國藥典(USP)和日本藥典(JP或JPE))專門用於製藥製劑的已知的聚乙烯醇不能或只有在特定條件下才能通過壓力作用直接壓片。
就此而言的一個特別的問題在於,如何以簡單方式製備主要由作為活性成分賦形劑的相應PVA組成的片劑,其中活性成分均勻分布。通常只有在較高比例的其它粘合劑(例如乳糖)和潤滑劑和可能的其它添加劑的存在下才能實現含PVA的製劑的可直接壓片性。通常在水溶液或醇溶液的存在下製備使用PVA作為活性成分載體的製劑。例如已知通過如下方式製備具有延長時間的活性成分釋放的相應片劑:在溼法造粒之後在其它摻加劑的存在下壓制活性成分和PVA。溼法造粒的缺點在於,必須通過使用大量能量重新除去溼法造粒所需的溶劑。
本發明的目的
通過上述內容可知,為了實現希望的緩釋效果,通常使用溶脹聚合物作為基質,例如在胃和腸中潤溼之後活性成分以時間受控的方式通過擴散過程和侵蝕過程從溶脹聚合物中釋放並且用於吸收。當例如活性成分與通常用作緩釋聚合物的羥丙基甲基纖維素(HPMC)之間存在不相容反應時或者當所使用的HPMC類型顯示出不令人滿意的活性成分釋放特性時,通常使用聚乙烯醇(PVA)。
為了迅速開發具有緩釋效果的片劑,製劑研究員需要可直接壓制並且以時間受控的方式釋放活性成分的溶脹聚合物。然而已知的粉末狀PVA本身不可直接壓制,它們形成不能在製藥實踐中使用的具有不足硬度的片劑,因為它們例如具有不希望的破碎傾向或過高的磨損。
因此為了迅速開發所述基於聚乙烯醇的緩釋片劑,希望可直接壓制的聚乙烯醇賦形材料。所述賦形材料使得無需進行為了使壓片混合物可壓制而通常必要的冗長和昂貴的造粒步驟。
因此本發明的目的是提供可直接壓制的緩釋基質,所述緩釋基質使得無需進行耗時的造粒方法;即無需進行例如由如下的步驟:使用造粒液體潤溼,在混合系統或流化床設備中機械混合,以及用於除去造粒液體的後乾燥方法和篩選,因此可以節省時間和能量,並且節省特殊造粒設備中的昂貴和耗時的投資。本發明的目的還在於,提供基於主要由PVA組成的組合物的所述有利的可直接壓制的緩釋基質。本發明的目的還在於,提供可以將PVA或市售PVA品質轉化成可直接壓制的狀態的方法。
本發明的簡要描述
本發明的主題是可直接壓制的共混物,所述共混物包含細粒聚乙烯醇(PVA)和細粒微晶纖維素(MCC),通過所述共混物可以提供具有緩釋的活性成分釋放的可直接壓制的蓋倫組合物。優選地,所述混合物為本發明的主題,其中所使用的細粒聚乙烯醇(PVA)和細粒微晶纖維素(MCC)滿足藥典(Ph.Eur.、USP/NF和JPE)的要求。特別通過可直接壓制的共混物實現本發明的目的的解決方案,所述共混物包含平均粒度Dv50<100μm,優選平均粒度Dv50<65μm,特別優選平均粒度Dv5072.2摩爾%)。
特別合適地,相應的可直接壓制的共混物包含根據USP通過下式表徵的作為水溶脹性樹脂的聚乙烯醇(PVA)
(C2H4O)n
其中
n表示500至5000範圍內的整數,
並且所述聚乙烯醇通過聚乙酸乙烯酯的85-89ige%的水解獲得。
此外,本發明的主題還有具有數小時的延長的活性成分釋放的含活性成分的片劑,更確切地說包含具有上述特徵的細粒聚乙烯醇(PVA)和細粒微晶纖維素(MCC)的共混物的片劑。
此外發現,基於片劑的總重量,以1-99重量%的量,優選以5-95重量%的量,非常特別優選以10-90重量%的量包含相應的可直接壓制的共混物的含活性成分的片劑具有希望的延長的活性成分釋放。
有利地,在使用低壓力時,藉助所述組合物已經可以獲得具有特別高的片劑硬度的片劑,所述片劑在製備過程中出人意料地需要低的推出力,並且僅具有≤0.2重量%的低脆碎度。
在使用根據本發明的共混物時,通過施加10kN的壓力已經可以獲得具有≥153N的片劑硬度和≤0.2重量%的脆碎度的片劑。在使用根據本發明的共混物時,通過以20kN的壓力壓制獲得具有≥289N的片劑硬度和≤0.1重量%的脆碎度的片劑。
使用所述共混物可以特別容易地製備具有延緩的活性成分釋放的片劑,所述片劑單獨或與其它活性成分組合地優選包含BCS類別I的活性成分。然而,如果希望並且存在臨床需要,也可以通過根據本發明的方法將其它BCS類別的活性成分加工成具有緩釋的活性成分釋放的可直接壓制的劑型。
本發明的詳細描述
藥劑的充分療效通常取決於均勻劑量和每天需要多次給藥,從而可以避免不希望的副作用。然而在患者依從性方面,每天多次給藥是不希望的。因此為了給藥一定的活性成分,希望提供片劑製劑,通過所述片劑製劑在數小時內緩慢地釋放活性成分,從而在定期服用時在一天內建立基本上恆定的有效血液水平,但是一天僅需要服用一次。
取決於待使用的活性成分,各種組合物的需要不同。根據待使用的活性成分的化學和物理性能,需要使用其它活性成分賦形劑和壓片助劑,因為並非每種活性成分都與每種壓片助劑相容,或者由於化學和物理性能而不能彼此加工。
可以根據美國九十年代中期由Gordon Amidon研發的生物藥劑分類系統(BCS)劃分活性成分的生物治療性,所述生物藥劑分類系統同時是US-FDA(食品和藥物管理局)指南以及歐洲藥品局指南的一部分,用於評價藥劑的生物等效性。
例如,BCS類別I的活性成分是具有高滲透能力的易溶性活性成分。其吸收僅受胃排空和腸排空的速度的控制。對於屬於該類別但是希望在一整天內產生療效的活性成分,嘗試研發能夠實現延緩且均勻的活性成分釋放的製劑。
生物藥劑分類系統(簡稱BCS,英文:Biopharmaceutics Classification System)描述了在口服藥物時起重要作用的關聯。所述系統基於G.Amidon和同事的1995年的論文。作者在該論文中描述了藥物的口服生物利用度主要受其溶解度、溶解速率以及通過生物膜的滲透性的影響(Amidon GL,Lennernas H,Shah VP;Crison JR.A theoretical basis for a biopharmaceutic drug Classification:the correlation of in vitro product dissolution and in vivo bioavailability.Pharm Res.1995;12:413.)。
對於BCS類別1的活性成分,溶解度和滲透性二者均是高的。
這意味著當藥物的溶解度和滲透性較高時,可以假設吸收速率主要由胃排空和腸排空的速度決定。
自從2000年8月以來,美國藥劑審批機關FDA(食品和藥物管理局)的審批流程中開始使用BCS系統。在一定前提下,在申請審批成品藥劑時,當使用BCS系統證實新的成品藥劑(FAM)和所述藥物的已經審批的FAM肯定生物等效時,可以無需進行生物利用度研究和生物等效性研究。之後可以申請放棄(英文:waiver)進行昂貴並且在該情況下不必要的生物利用度研究的義務。為此,藥物在各種藥物形式中必須滿足與主要參數溶解度、滲透性和溶解速率相關的一定要求。
溶解度:
最高劑量的藥物必須完全溶解在最多250mL的pH值範圍在pH 1和pH 7.5之間的含水溶解介質中。
滲透性:
當至少90%的給藥劑量被人體吸收時,藥物則具有高滲透性。這必須通過合適的數據加以證明(例如質量平衡研究)。
溶解速度:
藥物形式必須保證迅速的藥物釋放。這必須通過合適的體外釋放試驗(轉籃法或轉片法)加以證明。至少85%的相應劑量必須在30分鐘內在三種不同的釋放介質(0.1NHCL、pH 4.5緩衝液和pH 6.8緩衝液)中釋放。
如上所述,本發明的目標是提供在數小時內均勻的易溶性活性成分。出人意料地通過使用聚合的活性成分賦形劑實現了所述問題的解決方案,其中聚合的活性成分賦形劑在生理液體(例如唾液或胃液和腸液)的存在下緩慢地形成凝膠並且從片劑基質中受控地通過擴散緩慢釋放活性成分。
聚乙烯醇(PVA)適合於該目的,聚乙烯醇作為合成聚合物為水溶脹性樹脂並且具有出色的成膜性能和乳化性能,並且在水溶液中形成凝膠。PVA根據USP通過下式表徵
(C2H4O)n
其中
n表示500至5 000範圍內的整數。根據聚合物的分子尺寸及其化學組成,其性能(特別是關於水溶性以及可壓片性)大幅變化。PVA由聚乙酸乙烯酯製成,其中乙酸酯基團部分或完全水解從而獲得醇基團。隨著水解度的增加,聚合物在含水介質中的溶解度增加,但是聚合物的結晶度和熔點也增加。此外,玻璃化轉變溫度也依據水解度而變化。
例如,38%水解的材料不具有熔點,但是具有約48℃的玻璃化轉變溫度,相反75%水解的材料具有約178℃的熔點,88%水解的材料具有約196℃的熔點,並且99%水解的材料具有約220℃的熔點,但是其中聚合物傾向於在高於200℃的溫度下迅速分解。
為了製備根據本發明的組合物,可以使用18-88、26-88和40-88型聚乙烯醇(PVA)和品質介於其間的所有聚乙烯醇,包括根據JPE和Ph.Eur.的28-99型聚乙烯醇。
儘管聚乙烯醇溶於水,但除了少數溶劑之外(例如以低溶解度溶於乙醇)其幾乎不溶於幾乎所有有機溶劑。聚合物的所述性能使得非常難以製備包含高比例PVA並且可直接壓片的片劑製劑。
為了用在製藥製劑中,根據各種藥典列舉了具有不同水解度的聚乙烯醇。
歐洲藥典規定,用在製藥製劑中的經審批的聚乙烯醇必須具有不大於280的酯值和在20000和150000之間的平均相對分子量。水解百分比(H)可以通過如下方程計算:
H=((100-(0.1535)(EV))/(100-(0.0749)(EV)))x100,
其中EV對應於聚合物的酯值。酯值表示皂化1g樣品中的酯所需的氫氧化鉀量的單位為mg的數據。酯值通過皂化值和酸值的差值計算。
因此根據歐洲藥典的專題論文,僅可使用水解百分比大於72.2%的PVA聚合物。
根據美國藥典,適合用在製藥劑型中的聚乙烯醇必須具有在85和89%之間的水解度百分比和500至5000的聚合度。聚合度(DM)通過如下方程計算:
DM=(摩爾質量)/((86)-(0.42(水解度)))
根據歐洲藥典專題論文可以用在製藥製劑中的PVA為水解度在72.2%和90%之間的PVA,因此包括根據Ph.Eur.的PVA(水解度大於72.2%但是小於90%)和根據USP的PVA(水解度在85-89%之間)。這些PVA品質具有14000g/mol至250000g/mol範圍內的分子量。
目前通過試驗發現,對於片劑製劑的易加工性,不僅所使用的聚乙烯醇的水解度和因此結晶度起作用,而且所使用的市售PVA品質的物理性能和外觀形式也起作用。
如上所述,具有相應的高水解度的聚乙烯醇只有在特定條件下才能直接壓片,即其必須預先進行造粒步驟或者所使用的PVA必須與其它壓片助劑和易壓縮粘合劑混合,使得在整體組合物中的聚乙烯醇的比例降低。
目前通過試驗出人意料地發現,特別細粒的聚乙烯醇可以實現直接壓片性。當對用於製藥製劑的合適的聚乙烯醇進行研磨和篩選時,可以獲得相應的細粒聚乙烯醇。
通過這種方式可以製備包含PVA粉末的可直接壓片的混合物,其中可以出人意料高地設定PVA含量。
進行的試驗已表明,通過以合適的方式與其它聚合物助劑組合,這樣預處理的聚乙烯醇的壓片性可以得到進一步改進。亦即,經研磨的細粒粉末可以隨後與其它合適的聚合物助劑組合,由此進一步改進所獲得的共混物的壓制性。
在此發現,當經研磨的細粒聚乙烯與微晶纖維素混合時,獲得壓片性特別好的組合。為此可以使用被證明用於製藥製劑的市售微晶纖維素,例如JRS Pharma的102和類型以及FMCBiopolymer的PH 102類型。特別地,當所使用的微晶纖維素特別細粒時,顯示出共混物的明顯改進的壓制性。
對於可直接壓制的緩釋片劑的開發來說這是特別重要的,因為製劑研究員始終需要「更好的助劑」,即具有進一步改進的壓制性的基質。其原因在於,尋求的是能夠在直接壓片過程中加工可壓制性極差的API,然而使用具有低壓制性的DC材料不能實現這一目的。
此外在使用具有改進的壓制性的可直接壓制的壓片基質時,可以減少其用量,從而可以製備具有更小重量和減小尺寸的片劑,其中獲得的片劑也具有極好的片劑硬度(所謂的「稀釋效應」)。對於所謂的「高劑量」緩釋片劑來說,這些性能是特別引人注意的,因為減小的片劑尺寸改善了患者的吞咽,因此保證了依從性和由此的治療效果。
通過測試經研磨PVA類型與各種微晶纖維素(MCC)的壓片性的試驗出人意料地發現,可以根據所使用的MCC類型降低或提高壓制性。特別地,相比於其它MCC類型,類型Avicel PH105、Vivapur 101和Avicel PH101在相同的壓力下實現片劑硬度的明顯升高。這些MCC類型的詳細研究表明,其與其它類型的區別在於其粒度。MCC的粒度Dv50優選在17-67μm的範圍內。已經顯示,MCC粒度越精細,與細粒PVA組合實現的片劑硬度越好。因此優選地,用於製備根據本發明的共混物的MCC類型的粒度(以Dv50的形式通過雷射衍射測得)儘可能地小於100μm,特別優選平均粒度小於70μm,特別優選小於20μm。相反在使用「粗粒」(高達100μm,特別是高達180μm)時,片劑硬度明顯降低。
就此而言特別出人意料地發現,正如本研究表明,非常明顯只有所述MCC適合實現改進的可直接壓制的性能;其它常規促進直接壓制的賦形劑,例如可直接壓制的磷酸氫鈣(包括本身極易直接壓片的(日本Fuji Cemical Industry))、可直接壓制的山梨醇(例如Sl 400,德國Merck KGaA)、可直接壓制的甘露醇(例如M200,德國Merck KGaA)或可直接壓制的澱粉(例如澱粉1500,英國Colorcon Limited)在與PVA組合時不顯示該作用並且不能與PVA形成可直接壓制的粉末混合物。
通過所述出人意料發現的效果,製劑研究員目前可以提供用於製備片劑的主要由PVA和細粒微晶纖維素組成並且可直接壓制的預混物,這加速了新的片劑製劑的研發過程。
由於在直接壓制基質中在相同PVA/MCC比例下片劑硬度得到改進,製劑研究員目前能夠將迄今不能壓制或壓制性差的活性成分轉化成緩釋片劑。此外,製劑研究員目前還能夠將高劑量API轉化成具有無問題地吞咽尺寸的「患者友好」的緩釋片劑。此外對於相同的PVA量,目前能夠根據需要減少微晶纖維素的量,因此減小片劑重量和片劑尺寸而PVA的緩釋效果不會改變。相比於基於粗粒MCC類型的對比材料,這些材料造成更好的所謂的「稀釋」效應。
微晶纖維素(MCC)為製備藥劑的最重要的壓片助劑並且優選用作活性成分賦形劑,並且是幾乎所有類型的口服劑型(例如片劑、膠囊、袋劑、顆粒等)的重要組分。具有通式(C6H10O5)n的微晶纖維素(MCC)在純淨形式下為白色可自由流動的粉末狀纖維素,並且可以以不同粒度市售獲得。在製藥品質下,其滿足USP標準。微晶纖維素特別充當減少卡路裡的食品(例如沙拉醬、甜點和冰淇淋)的不可消化且不可吸收的纖維,充當隔離劑或賦形劑物質。如上所述,微晶纖維素在藥劑中充當用於製備片劑的粘合劑或賦形劑物質。在本文中,微晶纖維素被證明特別適合直接壓片並且導致對於合適的製劑具有短分解時間的硬質片劑。
MCC由木質植物部分獲得(並非來自廢紙)。在此,使用稀鹽酸在超過100℃的溫度下使植物纖維素不含非結晶的纖維素部分。亦即,能夠通過高純度纖維素的部分水解和之後的純化和乾燥獲得製藥品質的MCC。在水解之後可以任選進行羧化,從而改進親水性能。
MCC不溶於水、醇和有機溶劑。MCC在水中形成由大量不溶微晶組成的三維基質,所述微晶形成穩定的觸變凝膠。即使在由溫度造成的相態變化時(例如在轉變成凍結狀態時或者在加熱至較高溫時),MCC仍然保持其有利性能,使得MCC非常適合用於進一步加工的成品混合物。
被證明適合實現足夠片劑硬度的MCC是平均粒度Dv50(以Dv50的形式通過雷射衍射測得)儘可能小於100μm,優選小於70μm,特別優選Dv50在17-67μm範圍內,特別優選小於20μm的市售類型。在此,所述細粒MCC類型優選具有在0.20至0.35g/cm3範圍內,優選在0.20至0.31g/cm3範圍內的堆積密度。滿足這些標準並且有資格用在製藥製劑中的合適的市售MCC品質例如為Vivapur 101(在空氣流中乾燥,通過雷射衍射確定的平均粒度Dv50為65μm,堆積密度0.26-0.31g/cm3)、Avicel PH 101(平均粒度為50μm,堆積密度0.26-0.31g/cm3)以及Avicel PH 105(噴霧乾燥,通過雷射衍射確定的平均粒度Dv50為20μm,堆積密度0.20-0.30g/cm3)。
然而根據本文描述的發明也可以使用本文未提及的滿足本文描述的要求的其它市售產品。
特別出人意料的是,通過組合合適的微晶纖維素與各種PVA品質(特別是具有不同粘度的PVA),獲得可直接壓制的混合物,所述混合物根據需要主要由PVA組成,任選也可以由相同比例的PVA和微晶纖維素組成。當希望時,也可以使用細粒微晶纖維素的比例高於細粒聚乙烯醇的比例的混合物。
被證明特別有利的是,在根據本發明的組合物中,所述細粒聚乙烯醇和細粒微晶纖維素的基於重量的比例在5:1至1:5的範圍內,優選在2:1至1:2的範圍內,特別優選1:1。所述共混物被證明特別適合製備具有延緩的活性成分釋放的片劑。在密切混合之後,本文發現的PVA和MCC的共混物具有0.38-0.48g/ml範圍內的堆積密度和0.53-0.65g/ml範圍內的夯實密度。
由於細粒聚乙烯醇和細粒微晶纖維素的組合的所述有利性能,製劑研究員在製藥工業以及食品工業或其它技術領域中獲得材料,所述材料顯著簡化了具有延長的活性成分釋放的固體壓製劑型的研發工作。製劑研究員只需要混合待緩釋的活性成分和根據本發明的PVA-MCC組合,任選加入一些助劑(特別是潤滑劑)然後在壓片機上壓制所述混合物。由於所述基質的特別好的壓片性能,也能夠開發具有活性成分的緩釋片劑,所述緩釋片劑本身實際上不適合直接壓制並且必須在以常規方式進行的工藝中預先造粒。PVA-MCC基質的使用節省了研發時間和設備投資,並且導致在研發和生產中的改進的過程可靠性。
在壓片過程中,在使用根據本發明的共混物時,一個有利的副作用在於,根據本發明的混合物導致相對低的推出力,由此相比於壓片中通常使用的量,能夠使用明顯更少量的潤滑劑進行加工。因此代替通常使用的1%的硬脂酸鎂的加入量,只需要約四分之一的量,任選地甚至需要更少的量。在特定條件下還可以完全無需加入所述潤滑劑。這造成顆粒間結合力的額外改進,即在相同的壓力下獲得更硬的片劑,其中可以實現可重現的活性成分釋放。可重現的活性成分釋放的原因在於,釋放基本上受PVA含量的控制,加入更少的疏水性硬脂酸鎂僅對釋放行為產生輕微影響。
本發明還涉及用於影響僅具有低壓制性的細粒PVA品質(特別是經研磨PVA)的壓片性能的方法。通過試驗發現,細粒PVA通過與細粒MCC組合可以轉變成可直接壓制的形式。
細粒PVA特別適合用作緩釋基質,因為其通常極易使用從而製備具有待緩釋的活性成分的均勻混合物。為了在每個單獨片劑中始終獲得相同的活性成分量,均勻混合物對於單次劑量精確度含量均勻度摂來說具有重要意義。
此外具有細粒PVA品質的這種類型的製劑的優點在於,細PVA顆粒的較大表面造成在胃腸道中潤溼之後形成均勻的凝膠層,當患者服用片劑時這造成活性成分以可重現的並且還任選延長擴散通過凝膠。
工序
為了製備根據本發明的共混物,使經研磨的細粒聚乙烯醇(PVA)與所選擇的細粒微晶纖維素(MCC)密切混合併且轉化成共混物,所述共混物出色地適合作為可直接壓制的壓片基質。這是特別出人意料的,因為這樣的PVA與市面上的其它(本身也極易壓制的)可直接壓片的助劑的摻合物不能與粉末狀PVA(特別地也不能與任意微晶纖維素)顯示出所述直接壓制效果。只有當組合細粒PVA和特別細粒的微晶纖維素時,才能形成可直接壓制的共混物。
有利地,使用根據本發明的細粒共混物可以將壓制性較差的活性成分轉化成製劑,所述製劑可以非常容易地壓製成片劑而無需其它預處理。通過所製備的包含相應共混物作為活性成分賦形劑的片劑還顯示,這樣製備的片劑可以在極長的時間內以受控方式釋放活性成分。取決於所使用的活性成分和細粒聚乙烯醇與微晶纖維素的混合比例,相應的含活性成分的片劑顯示出至少2小時,優選超過至少6小時,特別優選至少8小時,特別優選至少10小時的延緩的活性成分釋放,非常特別優選長達12小時的活性成分釋放。
由於關於片劑製劑的製備並沒有以結合方式定義術語「可直接壓制」,在本說明書中使用市面上的極易壓制的甘露醇(M 200(甘露醇),適合用作賦形劑exp Ph.Eur,BP,JP,USP,E 421,貨號1.00419,Merck KGaA,達姆施塔特,德國)的壓制行為作為對比標準。目的是特別使包含更大量細粒PVA結合細粒微晶纖維素的可直接壓制的共混物在壓制性方面儘可能地接近M 200的行為。
通過進行的試驗發現,基於片劑的總重量,以共混物的形式以1-99重量%的量,優選以5-95重量%的量,非常特別優選以10-90重量%的量包含根據本發明的組合物的含活性成分的片劑具有希望的特別好的壓制性。有利地,在使用低壓力時,通過這樣的組合物正如希望的已經可以獲得具有特別高的片劑硬度的片劑,所述片劑在製備過程中出人意料地需要低的推出力。在使用20kN的壓力時已經可以獲得具有高達462N的片劑硬度的片劑,所述片劑僅需要小於60N的推出力。此外正如通過合適的試驗表明,所述片劑僅具有低的脆碎度。
因此,通過本發明提供可直接壓制的組合物的製備方法,所述組合物具有延長的活性成分釋放和特別好的壓制性,由此製備細粒微晶纖維素(MCC)和細粒聚乙烯醇(PVA)的共混物,其中將聚乙烯醇研磨成平均粒度Dv50在50至260μm範圍內、堆積密度在0.55至0.62g/ml範圍內並且休止角在35至38°範圍內的細粒粉末並且通過800μm的篩進行篩選,並且使所獲得的粉末與細粒微晶纖維素(MCC)混合,所述細粒微晶纖維素具有<100μm範圍內的平均粒度Dv50,優選DV50<70μm的平均粒度,特別優選17至67μm範圍內的平均粒度Dv50,特別是17μm-20μm範圍內的Dv50,並且具有0.20至0.35g/cm3範圍內,優選0.20至0.31g/cm3範圍內的堆積密度。通過這種方式獲得可直接壓制的共混物,所述共混物根據需要可以與不同的活性成分混合併且壓製成具有延緩的活性成分釋放的片劑。
下文給出的實施例公開了用於製備根據本發明的PVA-MCC共混物的方法和條件。對於本領域技術人員不言而喻的是,除了本文描述的方法之外,還可以使用其它方法來研磨和混合起始物質。
相比於通過細粒PVA與其它(也特別適合壓片的)賦形物質的組合獲得的不足的壓制性,通過實施例可知PVA-MCC組合的特別的優點。
在摻合根據本發明的細粒PVA-MCC基質和本身差壓制性的粉末狀抗壞血酸並且加入非常少量的硬脂酸鎂作為潤滑劑時,可以通過簡單的直接壓片獲得具有足夠硬度和低機械磨損的片劑,所述片劑可以無問題地進行進一步處理,例如裝入泡罩包裝或者被患者從推擠式包裝中無損壞的取出。相應的含活性成分的片劑表明,可以保證在數小時內從所述PVA-MCC基質片劑中延長地釋放活性成分。
附圖列表:
在附圖中,圖1至4以圖表方式顯示了試驗結果用於進行說明:
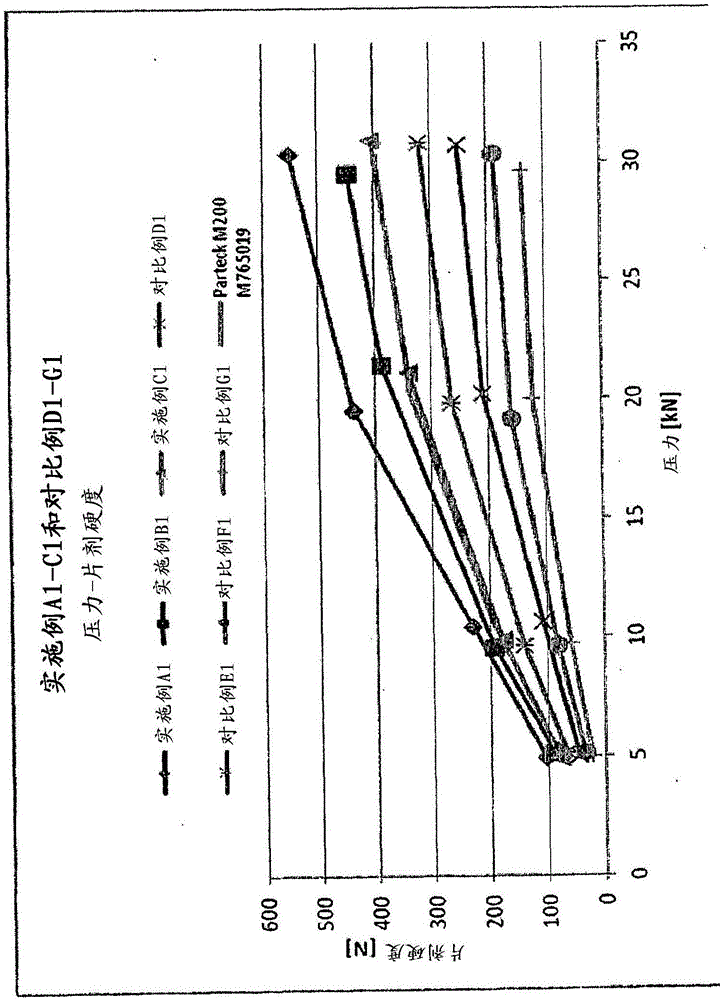
圖1:壓力-片劑硬度特性(得自表1b)
圖2:壓力-片劑硬度特性(得自表2b)
圖3:壓力-片劑硬度特性(得自表3b)
圖4:壓力-片劑硬度特性(得自表4b)
實施例
本說明書使得本領域技術人員能夠全面地實施本發明。雖然沒有其它論述,因此假設本領域技術人員可以在最寬的範圍內利用上述說明。
當描述不清楚時,不言而喻應當參見所引用的出版物和專利文獻。因此這些文獻被視為是本說明書的公開的一部分。
為了便於理解和說明本發明,下文給出實施例,所述實施例落入本發明的保護範圍內。實施例也用於說明可能的變體形式。然而由於所描述的發明原理的普遍適用性,實施例不用於縮小本申請的保護範圍。
此外,對於本領域技術人員不言而喻的是,在給出的實施例和其它說明中,組合物中包含的組分的量始終合計100重量%或100摩爾%,以整個組合物計,即使當通過給出的百分比範圍得出更高的值時也不能超過100重量%或100摩爾%。如果沒有另外聲明,百分比數據為重量%或摩爾%,除了以體積數據給出的比例之外。
在實施例和說明書以及權利要求書中給出的溫度以℃表示。
各個實施例中給出了根據本發明的特定PVA-MCC組合的製備條件。非常特別合適的是FMC Biopolymer公司的MCC類型Avicel PH 105(實施例A1-A4)和Avicel PH101(實施例C1-C4)以及JRS Pharma公司的Vivapur 101類型(實施例B1-B4)。在使用相當的壓力時通過這些材料獲得最硬的片劑,即這些特定組合顯示出最佳的「稀釋」潛力。
所使用的材料的表徵
1.所使用的PVA類型及其性能:
1.1待研磨原料
1.1.1.PVA 4-88,聚乙烯醇4-88,適用於賦形劑exp Ph.Eur.,USP,JPE,貨號1.41350,德國達姆施塔特Merck KGaA
1.1.2.PVA 18-88,聚乙烯醇18-88,適用於賦形劑exp Ph.Eur.,USP,JPE,貨號1.41355,德國達姆施塔特Merck KGaA
1.1.3.PVA 26-88,聚乙烯醇26-88,適用於賦形劑exp Ph.Eur.,USP,JPE,貨號1.41352,德國達姆施塔特Merck KGaA
1.1.4.PVA 40-88,聚乙烯醇40-88,適用於賦形劑exp Ph.Eur.,USP,JPE,貨號1.41353,德國達姆施塔特Merck KGaA
1.1.5.PVA 28-99,聚乙烯醇28-99,適用於賦形劑exp JPE,貨號1.41356,德國達姆施塔特Merck KGaA
這些PVA類型以數毫米大的粗顆粒的形式存在,其在所述形式下不能用作可直接壓制的壓片基質。
大顆粒不允許可重現地填充衝模,因此在(循環)壓片機的高旋轉速度下也不允許恆定的片劑重量。此外只有細粒PVA可以保證活性成分的均勻分布而不會出現在片劑中的分離效應。為了保證生產的每個片劑中的活性成分的單劑量精確度(含量均勻度),這是必不可少的。此外,只有通過細粒PVA才可以保證對於可重現緩釋來說必不可少的整個片劑本體中的均勻的凝膠形成。
出於這些原因,在用作可直接壓制的緩釋基質之前,對上述粗粒PVA類型必須進行粉碎,即研磨。
1.2經研磨PVA類型
1.2.1.經研磨PVA 4-88,獲自聚乙烯醇4-88,貨號1.41350
1.2.2.經研磨PVA 18-88,獲自聚乙烯醇18-88,貨號1.41355
1.2.3.經研磨PVA 26-88,獲自聚乙烯醇26-88,貨號1.41352
1.2.4.經研磨PVA 40-88,獲自聚乙烯醇40-88,貨號1.41353
1.2.5.經研磨PVA 28-99,獲自聚乙烯醇28-99,貨號1.41356
研磨:
在德國奧格斯堡公司Hosokawa Alpine的200AS型Aeroplex螺旋噴射磨機上使用液氮以低溫研磨的方式在0℃至負30℃下進行對PVA類型的研磨。所得的經研磨PVA類型的產品性能(特別是粉末特徵值例如堆積密度、夯實密度、休止角、BET表面積、BET孔體積以及粒度分布)列於下表:
堆積密度、夯實密度、休止角、BET表面積、BET孔體積:
(測量方法的細節參見如下方法)
*經研磨PVA
通過雷射衍射使用乾式分散(1bar背壓)確定的粒度分布:
單位為μm(測量方法的細節參見如下方法)
*經研磨PVA
通過雷射衍射使用乾式分散(2bar背壓)確定的粒度分布:
單位為μm(測量方法的細節參見如下方法)
*經研磨PVA
通過雷射衍射使用乾式分散(3bar背壓)確定的粒度分布:
單位為μm(測量方法的細節參見如下方法)
*經研磨PVA
通過雷射衍射使用溼式分散(在稀液矽油中)確定粒度分布:
單位為μm(測量方法的細節參見如下方法)
*經研磨PVA
通過塔式篩分確定粒度分布:
單位為重量百分比(測量方法的細節參見如下方法)
*經研磨PVA
2.用於製備具有聚乙烯醇(經研磨)的摻合物的微晶纖維素(MCC)
2.1PH 101,微晶纖維素,Ph.Eur.,NF,JP,美國FMC BioPolymer
2.2PH 102,微晶纖維素,Ph.Eur.,NF,JP,美國FMC BioPolymer
2.3PH 102SCG,微晶纖維素,Ph.Eur.,NF,JP,美國FMCBioPolymer
2.4PH 105,微晶纖維素,Ph.Eur.,NF,JP,美國FMC BioPolymer
2.5類型12,微晶纖維素,Ph.Eur.,NF,JP,德國羅森貝格JRS Pharma
2.6類型101,微晶纖維素,Ph.Eur.,NF,JP,德國羅森貝格JRS Pharma
2.7類型102Premium,微晶纖維素,Ph.Eur.,NF,JP,德國羅森貝格JRS Pharma
2.8類型200,微晶纖維素,Ph.Eur.,NF,JP,德國羅森貝格JRS Pharma
2.990M,微晶纖維素,Ph.Eur.,NF,JP,德國羅森貝格JRS Pharma
2.10LP 200,微晶纖維素,Ph.Eur.,NF,JP,德國羅森貝格JRS Pharma
2.11M 302,微晶纖維素,Ph.Eur.,NF,JP,BP,USP,臺灣MingtaiChemicalCo.Ltd.,
通過雷射衍射使用乾式分散(1bar背壓)確定的粒度分布:
單位為μm(測量方法的細節參見如下方法)
通過雷射衍射使用乾式分散(2bar背壓)確定的粒度分布:
單位為μm(測量方法的細節參見如下方法)
通過雷射衍射使用乾式分散(3bar背壓)確定的粒度分布:
單位為μm(測量方法的細節參見如下方法)
*經研磨PVA
通過雷射衍射使用溼式分散(在稀液矽油中)確定粒度分布:
單位為μm(測量方法的細節參見如下方法)
2.其餘材料
由於並沒有以結合方式定義術語「可直接壓制」,使用市面上的極易壓制的甘露醇的壓制行為作為標準:
M 200(甘露醇),適合用作賦形劑exp Ph.Eur.,BP,JP,USP,E 421,貨號1.00419,德國達姆施塔特Merck KGaA
目的是特別使可直接壓制的PVA的壓制性儘可能地接近M 200的行為。
用於表徵物質性能的設備/方法
1.堆積密度:根據DIN EN ISO 60:1999(德國版本)
-數據單位為「g/ml」
2.夯實密度:根據DIN EN ISO 787-11:1995(德國版本)
-數據單位為「g/ml」
3.休止角:根據DIN ISO 4324:1983(德國版本)
-數據單位為「度」
4.根據BET確定的表面積:根據S.Brunauer等人的文獻「BET Surface Area by Nitrogen Absorption」(Journal of American Chemical Society,60,9,1983)的評估和程序,設備:Micromeritics Instrument Corporation公司(美國)的ASAP 2420;氮氣;樣品重量:約3,0000g;加熱:50℃(5h);加熱速率3K/min;通過三次確定得到算術平均數據
5.通過雷射衍射使用乾式分散確定粒度:具有分散單元Scirocco 2000的Mastersizer 2000(英國Malvern Instruments Ltd.公司),在1、2和3bar的背壓下確定;Fraunhofer評估;分散劑RI:1.000,遮蔽極限:0.1-10.0%,託盤類型:一般目的,背景時間:7500毫秒,測量時間:7500毫秒,根據ISO 13320-1的程序以及來自設備生產商的技術手冊和說明書的信息;數據單位為體積%
6.通過雷射衍射使用溼式分散確定粒度:具有溼式分散單元Hydro 2000SM的Mastersizer 2000(英國Malvern Instruments Ltd.公司);分散介質:稀液矽油(生產商:德國Evonic Goldschmidt GmbH;生產商名稱:Tegiloxan3,生產商貨號:9000305);分散劑RI:1.403;攪拌速度:2500rpm;託盤類型:一般目的;背景時間:7500毫秒;測量時間:7500毫秒,遮蔽極限:7.0-13.0%;根據ISO 13320-1的程序以及來自設備生產商的技術手冊和說明書的信息;數據單位為體積%
工序:用稀液矽油填充懸浮室,按份加入樣品直至達到目標遮蔽範圍(7.0-13.0%),並且在2分鐘的等待時間之後開始測量。
7.通過乾式篩選經由篩選塔確定粒度:Retsch AS 200對照,Retsch公司(德國);物質量:約110.00g;篩選時間:30分鐘;振幅強度:1mm;間隔:5秒;根據DIN ISO 3310的具有金屬絲織物的分析篩;網孔寬度(單位μm):710、600、500、400、355、300、250、200、150、100、75、50、32;表格中的每個篩選粒度級的分布量的單位為「樣品重量的重量%」
8.以如下方式進行壓片試驗:
具有根據試驗部分給出的組成的混合物在封閉的不鏽鋼容器(容積:約2L,高度:約19.5cm,外部直徑:約12cm)中在實驗室滾筒混合器(瑞士Turbula T2A,Willy A.Bachofen公司)中混合5分鐘。
作為硬脂酸鎂,使用通過250μm篩的Parteck LUB MST(植物性硬脂酸鎂)EMPROVE exp Ph.Eur.,BP,JP,NF,FCC貨號1.00663(德國MerckKGaA)。
在具有評估系統Catman 5.0(德國Hottinger Baldwin Messtechnik–HBM公司)的儀表化偏心壓片機Korsch EK 0-DMS(德國Korsch公司)上壓製成500mg片劑(11mm衝模,圓形,平面,具有稜邊)。
根據試驗的壓力(標稱設置:~5、~10、~20和~30kN;有效測得的實際值在實施例中給出),製備至少100個片劑從而評估壓制數據並且確定蓋倫特徵值。
片劑硬度、直徑和高度:Erweka Multicheck 5.1(德國Erweka公司);對於每個壓力分別由20次片劑測量獲得的平均數據(算術平均值)。在製備片劑一天之後進行測量。
片劑磨損:脆碎度試驗設備TA420(德國Erweka公司);根據Ph.Eur.第7版「von nicht ü berzogenen Tabletten」的設備參數和測量程序。在製備片劑一天之後進行測量。
片劑質量:稱重20個片劑/壓力獲得平均值(算數平均值):具有天平Sartorius CPA 64(德國Sartorius公司)的Multicheck 5.1(德國Erweka公司)。在製備片劑一天之後進行測量。
試驗結果
通過試驗發現,特別地只有具有三種特定的微晶纖維素(MCC)的共混物才能實現良好的壓制性。
通過試驗還發現,顯然並非所有市售獲得的MCC類型在與經研磨PVA的共混物中顯示出壓制性的改進。
由於沒有以結合方式定義術語「可直接壓制」,使用市面上的極易壓制的甘露醇(M 200(甘露醇),適合用作賦形劑exp Ph.Eur.,BP,JP,USP,E 421,貨號100419,Merck KGaA,達姆施塔特,德國)的壓制行為作為標準。目的是特別使可直接壓制的PVA(以共混物的形式)在壓制性方面儘可能地接近M 200的行為。通過試驗發現,基於經研磨的細粒聚乙烯醇和細粒微晶纖維素(例如市售獲得的產物Avicel PH 105(實施例A1-A4)、Vivapur 101(實施例B1-B4)和Avicel PH101(實施例C1-C4))的共混物具有非常特別良好的壓制性。相比於被視為特別容易直接壓制的M200的壓制性,這些壓制性相當或甚至明顯更好。
這些特定的PVA-MCC共混物因此特別適合作為基質從而與差壓制性的活性成分組合在直接壓片中配製緩釋片劑。
步驟:
1a.
製備由市面上的各種微晶纖維素和經研磨PVA類型4-88的摻合物
1b.
壓制所述摻合物(加入0.25重量%的LUB MST)並且關於參數片劑硬度、片劑質量、片劑高度、片劑磨損以及必要推出力進行片劑表徵
2a.
製備由市面上的各種微晶纖維素和經研磨PVA類型18-88的摻合物
2b.
壓制所述摻合物(加入0.25重量%的LUB MST)並且關於參數片劑硬度、片劑質量、片劑高度、片劑磨損以及必要推出力進行片劑表徵
3a.
製備由市面上的各種微晶纖維素和經研磨PVA類型26-88的摻合物
3b.
壓制所述摻合物(加入0.25重量%的LUB MST)並且關於參數片劑硬度、片劑質量、片劑高度、片劑磨損以及必要推出力進行片劑表徵
4a.
製備由市面上的各種微晶纖維素和經研磨PVA類型40-88的摻合物
4b.
壓制所述摻合物(加入0.25重量%的LUB MST)並且關於參數片劑硬度、片劑質量、片劑高度、片劑磨損以及必要推出力進行片劑表徵
A)試驗結果:
1a.可直接壓制的賦形物質與經研磨PVA類型4-88的摻合物的製備
一般說明:使經研磨PVA 4-88穿過800μm的手篩從而除去可能的粗粒和團塊。稱重300g篩選的物料並且將其置入2L渦流混合容器,加入300g相應的表1a的微晶纖維素並且在渦流混合器T2A中混合5分鐘。
表1a:經研磨PVA 4-88與微晶纖維素的共混物的組成
*:經研磨PVA
1b.壓制摻合物並且進行片劑表徵
一般說明:在渦流混合容器中分別使498.75g如上製得的實施例A1-C1或對比例D1-G1的共混物與1.25g硬脂酸鎂混合,在渦流混合器T2A中再混合5分鐘並且在偏心壓機Korsch EK 0-DMS上壓片。
將1%LUB MST摻合的M200充當對比樣。備註:由於所得的極高推出力,因此在使用少量硬脂酸鎂的情況下不能壓制M200。
表1b:經研磨PVA 4-88與微晶纖維素的共混物的壓片數據
說明:
A:壓力[kN]
B:1天之後的片劑硬度[N]
C:片劑質量[mg]
D:片劑高度[mm]
E:磨損[%]
F:推出力(N)
圖1中以圖表方式顯示了明顯不同的壓力-片劑硬度特性用於進行更好的說明。
2a.可直接壓制的賦形物質與經研磨PVA類型18-88的摻合物的製備
一般說明:使經研磨PVA 18-88穿過800μm的手篩從而除去可能的粗粒和團塊。稱重300g篩選的物料並且將其置入2L渦流混合容器,加入300g相應的表2a的微晶纖維素並且在渦流混合器T2A中混合5分鐘。
表2a:經研磨PVA 18-88與微晶纖維素的共混物的組成
*:經研磨PVA
2b.摻合物的壓制和片劑表徵
一般說明:
在渦流混合容器中分別使498.75g如上製得的實施例A2-C2或對比例D2-G2的共混物與1.25g硬脂酸鎂混合,在渦流混合器T2A中再混合5分鐘並且在偏心壓機Korsch EK 0-DMS上壓片。
將1%LUB MST摻合的M200充當對比樣。備註:由於所得的極高推出力,因此在使用少量硬脂酸鎂的情況下不能壓制M200。
表2b:經研磨PVA 18-88與微晶纖維素的共混物的壓片數據
說明:
A:壓力[kN]
B:1天之後的片劑硬度[N]
C:片劑質量[mg]
D:片劑高度[mm]
E:磨損[%]
F:推出力(N)
圖2中以圖表方式顯示了明顯不同的壓力-片劑硬度特性用於進行更好的說明。
3a.可直接壓制的賦形物質與經研磨PVA類型26-88的摻合物的製備
一般說明:使經研磨PVA 26-88穿過800μm的手篩從而除去可能的粗粒和團塊。稱重300g篩選的物料並且將其置入2L渦流混合容器,加入300g相應的表3a的微晶纖維素並且在渦流混合器T2A中混合5分鐘。
表3a:經研磨PVA 26-88與微晶纖維素的共混物的組成
*:經研磨PVA
3b.壓制摻合物並且進行片劑表徵
一般說明:在渦流混合容器中分別使498.75g如上製得的實施例A3-C3或對比例D3-K3的共混物與1.25g硬脂酸鎂混合,在渦流混合器T2A中再混合5分鐘並且在偏心壓機Korsch EK 0-DMS上壓片。
將1%LUB MST摻合的M200充當對比樣。備註:由於在其他方面所得的極高推出力,因此在使用少量硬脂酸鎂的情況下不能壓制M200。
表3b:經研磨PVA 26-88與微晶纖維素的共混物的壓片數據
說明:
A:壓力[kN]
B:1天之後的片劑硬度[N]
C:片劑質量[mg]
D:片劑高度[mm]
E:磨損[%]
F:推出力(N)
圖3中以圖表方式顯示了明顯不同的壓力-片劑硬度特性用於進行更好的說明。
4a.可直接壓制的賦形物質與經研磨PVA類型40-88的摻合物的製備
一般說明:使經研磨PVA 40-88穿過800μm的手篩從而除去可能的粗粒和團塊。稱重300g篩選的物料並且將其置入2L渦流混合容器,加入300g相應的表4a的微晶纖維素並且在渦流混合器T2A中混合5分鐘。
表4a:經研磨PVA 40-88與微晶纖維素的共混物的組成
*:經研磨PVA
4b.壓制摻合物並且進行片劑表徵
一般說明:在渦流混合容器中分別使498.75g如上製得的實施例A4-C4或對比例D4-G4的共混物與1.25g硬脂酸鎂混合,在渦流混合器T2A中再混合5分鐘並且在偏心壓機Korsch EK 0-DMS上壓片。
與1%LUB MST混合的M200充當對比樣。備註:由於在其他方面所需的極高推出力,因此在使用少量硬脂酸鎂的情況下不能壓制M200。
表4b:經研磨PVA 40-88與微晶纖維素的共混物的壓片數據
說明:
A:壓力[kN]
B:1天之後的片劑硬度[N]
C:片劑質量[mg]
D:片劑高度[mm]
E:磨損[%]
F:推出力(N)
圖4中以圖表方式顯示了明顯不同的壓力-片劑硬度特性用於進行更好的說明。