有害生物防除劑的製造法的製作方法
2023-08-02 06:25:06 3
技術領域
本發明涉及具有2-醯基亞氨基吡啶結構的新的有害生物防除劑的製造方法。
現有技術
目前為止已發現多種有害生物防除劑,然而因為藥劑靈敏性低下的問題、效果的持續性、使用時的安全性等,目前依然追求新的藥劑。
尤其是在東亞、東南亞的水稻栽培中,如非專利文獻1所示,由對包含以吡蟲啉(Imidacloprid)為代表的新菸鹼(Neonicotinoids)類、及以氟蟲腈(Fipronil)為代表的苯基吡唑系藥劑等的主要殺蟲劑發展了藥劑抵抗性的飛蝨類造成的災害已表面化,期待針對發展了抵抗性的飛蝨類的特效藥。另外,要求穩定且廉價地提供作為有害生物防除劑所要求的量的這些新的藥劑。
作為具有2-醯基亞氨基吡啶結構的有害生物防除劑的製造方法,已知專利文獻1~3、非專利文獻2。專利文獻1公開了具有與後述式(I)所表示的化合物同樣的環結構的除草劑。專利文獻2、以及專利文獻3公開了具有與式(I)所表示的化合物類似的環結構的殺蟲劑。非專利文獻2公開了將與具有式(I)所表示的化合物類似的環結構的化合物作為合成中間體。
然而,專利文獻1、專利文獻2、專利文獻3、非專利文獻2所記載的製造方法,是將後述的式(Ba)所表示的化合物作為中間體的製造方法,沒有記載將後述的式(B)所表示的化合物作為中間體的製造。另外,專利文獻1、專利文獻2、專利文獻3、非專利文獻2中公開了將式(Ba)所表示的化合物作為中間體的製造方法,但沒有關於後述的式(Ia)所表示的化合物的製造的具體記載。另外,對於N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺,公開了結構式,作為該化合物的物性值記載了折射率為nD(25.5)=1.4818(專利文獻2的第1表化合物號3),但不包含在已認定有害生物防除活性的化合物組的列表中(專利文獻2的第2表、第3表)。
進而,專利文獻3公開了N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的結構式,作為該化合物的物性值記載了熔點為60~62℃(專利文獻3的表7的實施例號12),在實施例中並未舉出具有害生物防除活性的化合物的例子。專利文獻2及專利文獻3中並未公開N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的具體製造方法。
另外,非專利文獻3中公開了作為2-乙醯胺吡啶的互變異構體的N-(吡啶-2(1H)-亞基]-乙醯胺,但沒有記載其具體製造方法、和/或滷代衍生物的製造方法。
現有技術文獻
專利文獻
專利文獻1:歐洲專利申請公開第432600號說明書
專利文獻2:日本特開平05-78323號公報
專利文獻3:歐洲專利申請公開第268915號說明書
非專利文獻
非專利文獻1:Masaya Matsumura等,Pest Management Science,2008年、64卷、11號、1115~1121頁
非專利文獻2:Botho Kickhofen等,Chemische Berichte,1955年、88卷、1103~1108頁
非專利文獻3:Wladysl,aw Pietrzycki等,Bulletin des Societes Chimiques Belges,1993年、102卷、11-12號、709~717頁
技術實現要素:
發明要解決的課題
提供後述式(I)所表示的具有2-醯基亞氨基吡啶結構的有害生物防除劑,特別是提供穩定且廉價地提供作為有害生物防除劑所要求的量的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的製造方法。
用於解決課題的方法
即,根據第一發明,本發明者們通過以式(A)所表示的化合物作為出發物質,以式(B)所表示的化合物作為中間體,而得到作為目的有用的下述式(I)所表示的化合物,從而完成了本發明。
提供下述式(I)所表示的化合物的製造方法,
[式中,Ar表示可被取代的苯基、或可被取代的5~6元的雜環,R1表示可被取代的C1-6的烷基,Y表示氫原子、滷原子、羥基、可被滷原子取代的C1-6烷基、可被滷原子取代的C1-6烷氧基、氰基、甲醯基、硝基。]
該方法如下述反應式所示,包括:式(B)所表示的化合物的製造工序、和烷基化工序,
[式中,R1和Y表示與上述相同的含義,R2表示(1)三氟乙醯氧基、(2)可被滷原子取代的C1-6烷氧基、或苯基可被滷原子、甲基、氰基、硝基、甲氧基取代的苄氧基、(3)除了三氟乙醯氧基以外的可被滷原子取代的C1-6烷基羰氧基、苯基可被滷原子、甲基、氰基、硝基、甲氧基取代的苯基羰氧基、(4)羥基、(5)滷原子,R4表示滷原子、可被滷原子取代的C1-6烷基磺醯氧基(C1-6alkylsulfoxy group)、可被滷原子或甲基取代的苯基磺醯氧基(phenylsulfoxy group)。
所述式(B)所表示的化合物的製造工序是,將式(A)所表示的化合物的2位氨基,使用R1COR2所表示的醯基化劑進行醯基化,從而製造式(B)所表示的化合物的工序,
所述烷基化工序是,將式(B)所表示的化合物的1位氮原子進一步使用Ar-CH2-R4進行烷基化的工序。
根據第二發明,提供所述式(B)所表示的有用中間體(但是,R1為甲基或苯基且Y為氫原子的化合物除外)、及其鹽。
根據第三發明,提供下述式(Ia)所表示的化合物的製造方法,
[式中,R3表示滷原子、氰基、硝基、三氟甲基,X表示碳原子或氮原子,R1a表示被滷素取代的C1-6烷基。]
該製造方法如下述反應式所示。
[其中,R1a、R4、R3及X表示與上述相同的含義,R2a表示(1)三氟乙醯氧基、(2)可被滷原子取代的C1-6烷氧基、或苯基可被滷原子、甲基、氰基、硝基、甲氧基取代的苄氧基、(3)除了三氟乙醯氧基以外的可被滷原子取代的C1-6烷基羰氧基、苯基可被滷原子、甲基、氰基、硝基、甲氧基取代的苯基羰氧基、(4)羥基、或(5)滷原子。]
根據第四發明,提供通過下述反應式所製造的式(I』)所表示的化合物。
[其中,式(I』)所表示的化合物是具有以下的(a)或(b)、或者(a)及(b)的物性的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺。(a)在粉末X射線衍射中,至少在下述衍射角(2θ)具有衍射角峰。衍射角:8.6±0.2°、14.2±0.2°、17.5±0.2°、18.3±0.2°、19.7±0.2°、22.3±0.2°、30.9±0.2°、35.3±0.2°
(b)在差示掃描量熱分析(DSC)中,熔點顯示155~158℃。]
發明的效果
通過本發明,可以根據需要通過一鍋(One-pot)有效且收率良好地製造作為有害生物防除劑有用的2-醯基亞氨基吡啶衍生物。
附圖說明
圖1是顯示以第一製法調製的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的晶體的粉末X射線晶體分析的結果的圖。
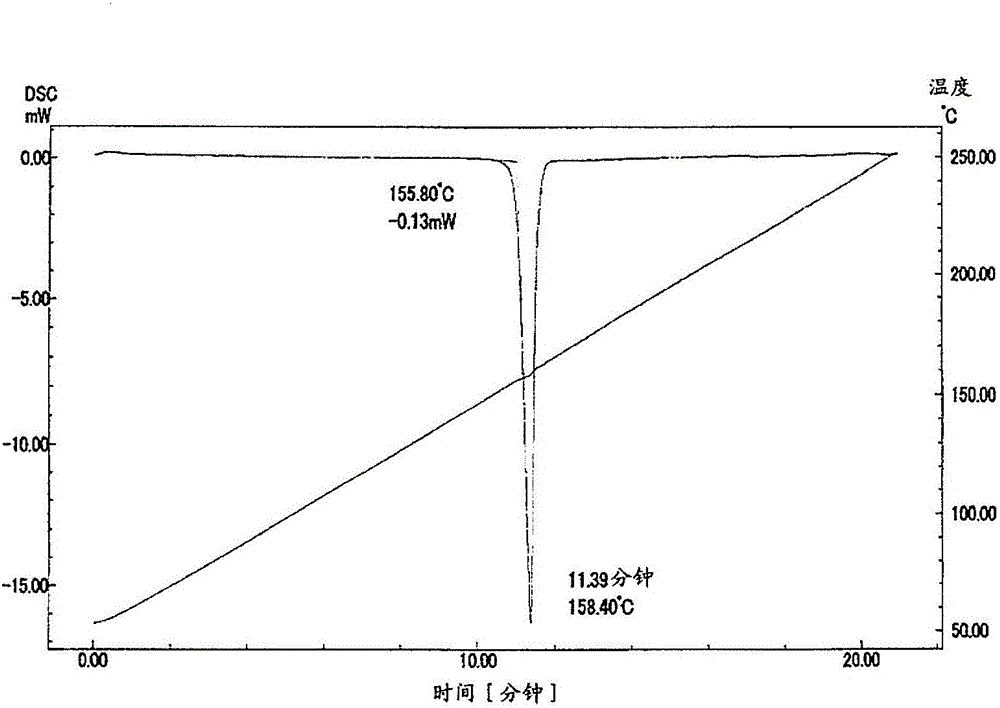
圖2是顯示以第一製法調製的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的晶體的差示掃描量熱分析的結果的圖。
圖3是顯示以第二製法調製的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的晶體的粉末X射線晶體分析的結果的圖。
圖4是顯示以第二製法調製的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的晶體的差示掃描量熱分析的結果的圖。
圖5是顯示以第三製法調製的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的晶體的差示掃描量熱分析的結果的圖。
圖6是顯示以第四製法調製的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的晶體的粉末X射線晶體分析的結果的圖。
圖7是顯示以第四製法調製的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的晶體的差示掃描量熱分析的結果的圖。
圖8是顯示以第五製法調製的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的晶體的差示掃描量熱分析的結果的圖。
圖9是顯示合成例4中合成的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的晶體的粉末X射線晶體分析的結果的圖。
具體實施方式
在本說明書中,作為取代基或取代基的一部份的「烷基」這樣的用語,只要沒有特別定義,則分別是指直鏈狀、支鏈狀、環狀或它們的組合的烷基。
在本說明書中,「滷原子」是指選自氟、氯、溴、碘中的原子。
在本說明書中,作為鹼的「當量」,例如相對於1摩爾式(A)所表示的化合物使用1摩爾碳酸鉀的情形時為2當量,使用1摩爾氫氧化鈉或碳酸氫鈉的情形時為1當量,使用1摩爾有機鹼的情形時為1當量。
在本說明書中,「鹽」表示鹽酸鹽、硫酸鹽、硝酸鹽等無機酸鹽、三氟乙酸鹽、二氟乙酸鹽、二氯乙酸鹽等有機酸鹽等。
在本說明書中,R2表示羥基時,與醯基化劑R1COR2同時使用的「試劑」可以是其水合物。
在本說明書中,「縮合劑」表示N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽、1,1』-羰基二咪唑、二吡啶基二硫、二咪唑基二硫、1,3,5-三氯苯甲醯氯、1,3,5-三氯苯甲酸酐、PyBop(註冊商標,六氟磷酸(苯並三唑-1-基氧基)三吡咯烷基)、PyBrop(註冊商標,六氟磷酸溴三(吡咯烷基))等用於合成酯和/或醯胺等羧酸衍生物的反應試劑。
在本說明書中,取代基所附帶的「Ca-b」記號,是指該取代基所含有的碳原子數為a個~b個。另外,例如,在「Ca-b烷基羰基氧基」這種情形下的「Ca-b」,是指除了羰基氧基部分的碳原子之外,烷基部分的碳原子數為a個~b個。
Ar表示可被取代的苯基、或可被取代的5~6元的雜環,作為5~6元的雜環,可列舉吡啶、嘧啶、噻唑、四氫呋喃、呋喃等,優選列舉3-吡啶基、5-嘧啶基、3-噻唑基、5-噻唑基,更優選為3-吡啶基。作為可被導入到所述苯基或雜環中的取代基,可列舉滷原子、可被滷原子取代的C1-4烷基、可被滷原子取代的C1-4烷氧基、羥基、氰基、硝基,優選為滷原子、可被滷原子取代的C1-4烷基,特別優選為氯原子。作為可被取代的苯基、及可被取代的5~6元的雜環,具體為苯基、3-氯苯基、4-氯苯基、3-氰基苯基、4-氰基苯基、3-硝基苯基、4-硝基苯基、3,5-二氯苯基、4-甲基苯基、4-甲氧基苯基、3,5-二溴苯基、2,4-二溴苯基、4-氟苯基、4-溴苯基、3-硝基-5-溴苯基、3,5-雙三氟甲基苯基、6-氯-3-吡啶基、2-氯-5-噻唑基、6-氯-5-氟-3-吡啶基、6-溴-3-吡啶基、6-氟-3-吡啶基、5,6-二氯-3-吡啶基、6-三氟甲基-3-吡啶基,優選為6-氯-3-吡啶基、6-氟-3-吡啶基、6-氯-5-氟-3-吡啶基、6-溴-3-吡啶基,特別優選為6-氯-3-吡啶基。
R1表示可被取代的C1-6烷基,可被導入到所述C1-6烷基中的取代基可列舉滷原子、C1-6滷代烷氧基、氰基、硝基、羥基,具體表示三氟甲基、二氟氯甲基、三氯甲基、五氟乙基、二氟甲基、二氯甲基、二溴甲基、氯甲基、二氟乙基、二氯乙基、2,2,2-三氟乙基、二氟環丙基、溴二氟甲基、三氟甲氧基甲基等,優選表示三氟甲基、二氟氯甲基、二氟甲基、三氯甲基、五氟乙基,更優選表示三氟甲基。
R1a表示可被滷素取代的C1-6烷基,可列舉三氟甲基、三氯甲基、二氟氯甲基、二氟甲基、二氯甲基、二溴甲基、氯甲基、二氟乙基、二氯乙基、2,2,2-三氟乙基、五氟乙基、二氟環丙基等,優選為三氟甲基、三氯甲基、二氯甲基、二氟甲基、二氟氯甲基、氯甲基、五氟乙基,更優選為三氟甲基、二氟甲基、二氟氯甲基、氯甲基、五氟乙基,特別優選為三氟甲基。
Y表示氫原子、滷原子、羥基、可被滷原子取代的C1-6烷基、可被滷原子取代的C1-6烷氧基、氰基、甲醯基、硝基,優選表示氫原子、滷原子、羥基,更優選表示氫原子。
R2及R2a表示(1)三氟乙醯氧基、(2)可被滷原子取代的C1-6烷氧基、或苯基可被滷原子、甲基、氰基、硝基、甲氧基取代的苄氧基、(3)除了三氟乙醯氧基以外的可被滷原子取代的C1-6烷基羰氧基、苯基可被滷原子、甲基、氰基、硝基、甲氧基取代的苯基羰氧基、(4)羥基、或(5)滷原子。
R3表示在吡啶環或嘧啶環的碳原子上取代的取代基,明顯其數在吡啶的情況下為0~4,在嘧啶的情況下為0~3。作為R3所表示的取代基,為滷原子、氰基、硝基、三氟甲基,可以各自相同也可以不同。
R4表示滷素、可被滷原子取代的C1-6烷基磺醯氧基、可被滷原子、甲基取代的苯基磺醯氧基。
作為式(I)及(Ia)所表示的化合物,優選為化合物號1:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺、化合物號2:N-[1-((6-氯-5-氟吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺、化合物號19:N-[1-((6-氟吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺、化合物號3:N-[1-((6-溴吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺、化合物號8:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2-二氟乙醯胺、化合物號4:2-氯-N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2-二氟乙醯胺、化合物號7:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,3,3,3-五氟丙醯胺、化合物號6:N-[1-((2-氯嘧啶-5-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺。
在式(I)及式(Ia)所表示的化合物中,作為特別優選的例子,是式(I』)所表示的化合物,即,具有以下(a)或(b)、或者(a)及(b)的物性的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺(但是,除了專利文獻2所記載的顯示nD(25.5)=1.4818的化合物之外)。
(a)在粉末X射線衍射中,至少在下述衍射角(2θ)具有衍射角峰。衍射角:8.6±0.2°、14.2±0.2°、17.5±0.2°、18.3±0.2°、19.7±0.2°、22.3±0.2°、30.9±0.2°、35.3±0.2°
(b)在差示掃描量熱分析(DSC)中,熔點顯示155~158℃。
作為式(B)所表示的化合物的優選例,可列舉2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺、2-氯-2,2-二氟-N-(吡啶-2(1H)-亞基)乙醯胺、2,2,3,3,3-五氟-N-(吡啶-2(1H)-亞基)丙醯胺、2,2-二氟-N-(吡啶-2(1H)-亞基)乙醯胺,作為更優選的例子是下式(B1)所表示的2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺。
製造方法
按照以下的流程圖更詳細地說明本發明。
[在上述的流程圖中,Ar、Y、R1、R2、R4表示與上述相同的含義。]
另外,上述流程圖所示的式(B)所表示的化合物可以不進行後處理或分離地在之後的工序中使用。
1-1:從式(A)所表示的化合物製造式(B)所表示的化合物
式(A)所表示的化合物可以是市售的化合物或通過例如Journal of labeled compounds&radiopharmaceuticals(1987),24(2)119-123所記載的方法獲得。
作為從式(A)所表示的化合物製造式(B)所表示的化合物的方法,可以通過在無溶劑條件下或在不影響反應的溶劑中,使式(A)所表示的化合物,在鹼存在下或不存在下,與醯基化劑R1COR2(R1、R2表示與上述所定義的相同的含義)反應而得到。
這裡所說的試劑的當量數是相對於全部式(A)所表示的化合物的當量數。
作為可用的溶劑,可列舉甲苯、二甲苯、乙苯等芳香族烴系溶劑、乙酸乙酯、乙酸丁酯等酯系溶劑、乙醚、異丙醚、四氫呋喃、二烷等醚系溶劑、N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、N-甲基-2-吡咯烷酮、乙腈等非質子性極性有機溶劑、二氯甲烷、氯仿等滷素系溶劑、環己烷等烴系溶劑、丙酮、甲乙酮等酮系溶劑、水、或它們的混合溶劑。
作為可用的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鈉、碳酸氫鉀、氫氧化鈉、氫氧化鎂、氫氧化鈣、氫氧化鋰、氫氧化鋇等無機鹼、1,8-二氮雜二環[5.4.0]十一碳-7-烯、1,5-二氮雜二環[4.3.0]壬-5-烯、三乙胺、二異丙基乙基胺、吡啶、甲基吡啶、二甲基氨基吡啶等有機鹼、乙醇鈉、甲醇鈉、叔丁醇鉀等醇化物。鹼也可以不使用,但在鹼存在下進行時的使用量,可用0.01~20.0當量。
作為醯基化劑R1COR2,可列舉三氟乙酸酐、三氟乙酸、三氟乙酸乙酯、三氟乙醯氯、或混合酸酐。另外,這樣的醯基化劑可以單獨使用或2種以上混合使用。其中,優選可使用三氟乙酸酐、三氟乙酸、三氟乙酸乙酯、或三氟乙醯氯。另外,R2表示羥基時,可以通過將N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽、1,1』-羰基二咪唑、二吡啶基二硫、二咪唑基二硫、1,3,5-三氯苯甲醯氯、1,3,5-三氯苯甲酸酐、PyBop(註冊商標,六氟磷酸(苯並三唑-1-基氧基)三吡咯烷基)、PyBrop(註冊商標,六氟磷酸溴三(吡咯烷基))等縮合劑、五氧化二磷、硫酸、多聚磷酸、亞硫醯氯、三氯氧磷、乙二醯氯、三氟化硼、對甲苯磺酸、或鐵、鈷、銅、鎳、鋅、鋁、鋰或鎂的滷化物、硫酸鹽、硝酸鹽或氧化物等試劑同時使用來進行。另外,這些試劑可單獨使用或2種以上組合使用。作為鐵、鈷、銅、鎳、鋅、鋁、鋰或鎂的滷化物、硫酸鹽、硝酸鹽或氧化物的優選例,可列舉氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁。另外,這些金屬化合物可以是其無水合物,也可以是水合物。醯基化劑的使用量優選為0.5~10.0當量,更優選為1.0~5.0當量。
反應溫度優選為-80℃~200℃的範圍。反應時間優選為0.1小時~7天的範圍。
作為優選的方式,
(1)在R2表示三氟乙醯氧基時,具體地在作為醯基化劑使用三氟乙酸酐時,作為優選的溶劑,可列舉乙酸乙酯、乙酸丁酯等酯系溶劑、二氯甲烷、氯仿等滷素系溶劑、甲苯、二甲苯、乙苯等芳香族烴系溶劑,更優選為甲苯。反應優選在鹼不存在下進行,但在使用鹼的情況下,作為優選的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鉀、三乙胺、吡啶等,更優選為碳酸鉀。醯基化劑的使用量優選為1.0~5.0當量,更優選為1.0~1.5當量。使用鹼時的鹼使用量優選為1.0~4.5當量,更優選為1.0~3.0當量。反應溫度優選為-20℃~50℃的範圍,更優選為-10℃~30℃。反應時間優選為0.1小時~7天的範圍,更優選為0.5小時~4小時的範圍。特別優選的條件是,作為醯基化劑使用三氟乙酸酐,作為溶劑使用甲苯,醯基化劑的使用量為1.0~1.5當量,反應溫度為-10℃~30℃,反應時間為0.5~4小時的條件。鹼不存在下,或在使用鹼的情況下是使用碳酸鉀、其當量數使用1.0~3.0當量的條件。
(2)在R2表示可被滷原子取代的C1-6烷氧基、或苯基可被滷原子、甲基、氰基、硝基、甲氧基取代的苄氧基時,具體地在使用三氟乙酸乙酯、三氟乙酸甲酯、三氟乙酸丙酯、特別優選為三氟乙酸乙酯等時,作為優選的溶劑,為N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、N-甲基-2-吡咯烷酮、乙腈等非質子性極性有機溶劑、乙醚、異丙醚、四氫呋喃、二烷等醚系溶劑、及這些溶劑與甲苯、二甲苯、乙苯等芳香族烴系溶劑的混合溶劑,優選為N,N-二甲基甲醯胺、或N,N-二甲基甲醯胺與甲苯的混合溶劑。反應優選在鹼不存在下進行,但在使用鹼的情況下,作為優選的鹼,可列舉碳酸鉀、三乙胺、二甲基氨基吡啶等,更優選為碳酸鉀、二甲基氨基吡啶。醯基化劑的使用量優選為1.0~5.0當量,更優選為1.5~5.0當量。使用鹼時的鹼使用量優選為0.01~3.0當量,更優選為0.01~2.0當量。反應溫度優選為20℃~100℃的範圍,更優選為40℃~80℃。反應時間優選為0.1小時~7天的範圍,更優選為1小時~2天的範圍。
特別優選的條件是,作為醯基化劑使用三氟乙酸乙酯,作為溶劑使用N,N-二甲基甲醯胺、或N,N-二甲基甲醯胺與甲苯的混合溶劑,醯基化劑的使用量為1.5~5.0當量,反應溫度為40℃~80℃,反應時間為2小時~2天的條件。鹼不存在下,或在使用鹼的情況下是使用碳酸鉀、其當量數使用1.0~3.0當量的條件。
(3)在R2表示可被滷原子取代的C1-6烷基羰氧基(但是,除了三氟乙醯氧基以外)、苯基可被滷原子、甲基、氰基、硝基、甲氧基取代的苯基羰氧基時,具體可列舉三甲基乙醯基。反應溫度優選為-20℃~50℃的範圍,更優選為-10℃~30℃。反應時間優選為0.1小時~7天的範圍,更優選為0.5小時~4小時的範圍。
(4)在R2表示羥基時,作為具體的醯基化劑可列舉三氟乙酸、二氟氯乙酸、三氯乙酸、二氟乙酸、二氯乙酸、二溴乙酸、氯乙酸、二氟丙酸、二氯丙酸、2,2,2-三氟丙酸、五氟丙酸、二氟環丙烷甲酸等,優選為三氟乙酸、三氯乙酸、二氯乙酸、二氟乙酸、二氟氯乙酸、氯乙酸、五氟丙酸,更優選為三氟乙酸、二氟乙酸、二氟氯乙酸、五氟丙酸,特別優選為三氟乙酸。使用三氟乙酸時,作為優選的溶劑,可列舉甲苯、二甲苯、乙苯等芳香族烴系溶劑及N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、N-甲基-2-吡咯烷酮、乙腈等非質子性極性有機溶劑,更優選為甲苯、二甲苯、N,N-二甲基甲醯胺、N-甲基-2-吡咯烷酮、N,N-二甲基乙醯胺、甲苯與N,N-二甲基甲醯胺的混合溶劑、二甲苯與N,N-二甲基甲醯胺的混合溶劑、二甲苯與N-甲基-2-吡咯烷酮的混合溶劑或二甲苯與N,N-二甲基乙醯胺的混合溶劑。作為同時使用的試劑,可列舉N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽、五氧化二磷、硫酸、多聚磷酸、亞硫醯氯、三氯氧磷、乙二醯氯等,這些溶劑優選使用0.2~5.0當量。另外,在作為同時使用的試劑使用氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁、三氟化硼、對甲苯磺酸等時,優選使用0.0001~1.0當量。反應使用五氧化二磷、硫酸、多聚磷酸、亞硫醯氯、三氯氧磷、乙二醯氯、氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁、三氟化硼、對甲苯磺酸時,優選在鹼不存在下進行,使用N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽時,優選在鹼存在下進行。使用鹼的情況下,作為優選的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鉀、三乙胺、吡啶、二甲基氨基吡啶等,更優選為三乙胺。醯基化劑的使用量優選為1.0~5.0當量,更優選為1.0~3.0當量。使用亞硫醯氯、三氯氧磷、乙二醯氯時,這些試劑使用0.2~5.0當量,反應溫度優選為-30℃~80℃的範圍,更優選為-10℃~40℃。使用五氧化二磷、硫酸、多聚磷酸時,這些試劑使用0.2~5.0當量,反應溫度優選為-30℃~200℃的範圍,更優選為-10℃~160℃。使用N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽時,優選為這些試劑使用0.2~5.0當量,反應溫度優選為-30℃~80℃的範圍,更優選為-10℃~40℃,以三乙胺作為鹼,使用0.2~5.0當量的條件。使用氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁、三氟化硼、對甲苯磺酸時,這些試劑使用0.0001~1.0當量,反應溫度優選為20℃~200℃的範圍,更優選為80℃~160℃。反應時間優選為0.1小時~7天的範圍,更優選為0.5小時~2天的範圍。
特別優選的條件是,作為醯基化劑使用三氟乙酸,作為溶劑使用甲苯、N,N-二甲基甲醯胺、二甲苯、N-甲基-2-吡咯烷酮、N,N-二甲基乙醯胺、N,N-二甲基甲醯胺與甲苯的混合溶劑、N,N-二甲基甲醯胺與二甲苯的混合溶劑、二甲苯與N-甲基-2-吡咯烷酮的混合溶劑或二甲苯與N,N-二甲基乙醯胺的混合溶劑,醯基化劑的使用量為1.0~3.0當量。使用亞硫醯氯、三氯氧磷、乙二醯氯時的特別優選的條件是,這些試劑使用0.3~3.0當量,在鹼不存在下,反應溫度為-10℃~40℃,反應時間為0.5小時~1天的條件。使用五氧化二磷、硫酸、多聚磷酸時的特別優選的條件是,這些試劑使用0.2~2.0當量,反應溫度為-10℃~160℃,反應時間為0.5小時~1天的條件。使用N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽時的特別優選的條件是,這些試劑使用0.5~3當量,反應溫度為-10℃~40℃,以三乙胺作為鹼,使用0.5~3.0當量,反應時間為0.5~1天的條件。使用氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁、三氟化硼、對甲苯磺酸時的特別優選的條件是,這些試劑使用0.0001~0.5當量,在鹼不存在下,反應溫度為80℃~160℃,反應時間為2小時~2天的條件。
(5)在R2表示滷原子時,具體在使用三氟乙醯氯、三氟乙醯溴,優選為三氟乙醯氯時,作為優選的溶劑,可列舉氯仿、二氯甲烷等的滷素系溶劑、甲苯、二甲苯、乙苯等芳香族烴系溶劑、及N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、N-甲基-2-吡咯烷酮、乙腈等非質子性極性有機溶劑,更優選為甲苯、N,N-二甲基甲醯胺、N-甲基-2-吡咯烷酮、或它們的混合溶劑。反應優選在鹼不存在下進行,但在使用鹼的情況下,作為優選的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鉀、三乙胺、吡啶等,更優選為碳酸鉀。醯基化劑的使用量優選為1.0~5.0當量,更優選為1.0~3.0當量。使用鹼時的鹼使用量優選為1.0~5.0當量,更優選為1.0~3.0當量。反應溫度優選為-80℃~40℃的範圍,更優選為-30℃~30℃。反應時間優選為0.1小時~7天的範圍,更優選為0.5小時~8小時的範圍。另外,R2表示氯原子時,還可以使用將三氟乙酸與亞硫醯氯、三氯氧磷、乙二醯氯等,在與式(A)所表示的化合物反應的體系外同時使用從而預先生成的R1COCl。
特別優選的條件是,作為醯基化劑使用三氟乙醯氯,作為溶劑使用甲苯、N,N-二甲基甲醯胺、N-甲基-2-吡咯烷酮或它們的混合溶劑,醯基化劑的使用量為1.0~3.0當量,反應溫度為-30℃~30℃,反應時間為0.5小時~8小時的條件。特別優選鹼不存在下,或在使用鹼的情況下使用碳酸鉀、其當量數使用1.0~3.0當量的條件。
在從式(A)所表示的化合物合成式(B)所表示的化合物後,可使用鹼進行中和。作為可用的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鈉、碳酸氫鉀、氫氧化鈉、氫氧化鎂、氫氧化鈣、氫氧化鋰、氫氧化鋇等無機鹼、1,8-二氮雜二環[5.4.0]十一碳-7-烯、1,5-二氮雜二環[4.3.0]壬-5-烯、三乙胺、二異丙基乙基胺、吡啶、甲基吡啶、二甲基氨基吡啶等有機鹼、乙醇鈉、甲醇鈉、叔丁醇鉀等醇化物,優選為碳酸鉀、乙醇鈉、三乙胺。
1-2:從式(B)或式(B』)所表示的化合物製造式(I)或式(I』)所表示的化合物
作為從式(B)或式(B』)所表示的化合物製造式(I)或式(I』)所表示的化合物的方法,可以通過在無溶劑條件下或在不影響反應的溶劑中,使式(B)或式(B』)所表示的化合物在鹼存在下與Ar-CH2-R4(Ar、R4表示與上述所定義的相同的含義)反應而得到。
作為可用的溶劑,可列舉乙醚、異丙醚、四氫呋喃、二烷等醚系溶劑、N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、乙腈、N-甲基-2-吡咯烷酮、N-甲基-2-哌嗪酮、N,N-二甲基-2-咪唑烷酮、乙腈等非質子性極性有機溶劑、二氯甲烷、氯仿等滷素系溶劑、甲苯、二甲苯、乙苯等芳香族烴系溶劑、及它們的混合溶劑,優選列舉非質子性極性有機溶劑。更優選為N,N-二甲基甲醯胺、N,N-二甲基乙醯胺、N-甲基-2-吡咯烷酮、N,N-二甲基-2-咪唑烷酮、乙腈,或N,N-二甲基甲醯胺、N,N-二甲基乙醯胺、N-甲基-2-吡咯烷酮、N,N-二甲基-2-咪唑烷酮、乙腈與芳香族烴系溶劑的混合溶劑,特別優選為N,N-二甲基甲醯胺、或N,N-二甲基甲醯胺與甲苯的混合溶劑。
作為在鹼存在下進行時的可用的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鈉、碳酸氫鉀、氫氧化鈉、氫氧化鎂、氫氧化鈣、氫氧化鋰、氫氧化鋇等無機鹼、1,8-二氮雜二環[5.4.0]十一碳-7-烯、1,5-二氮雜二環[4.3.0]壬-5-烯、三乙胺、二異丙基乙基胺、吡啶、二甲基吡啶、可力丁、N,N-二甲基苯胺、N,N-二乙基苯胺等有機鹼,優選列舉碳酸鉀、碳酸氫鉀、吡啶、三乙胺等,更優選列舉碳酸鉀、三乙胺。
Ar-CH2-R4(Ar、R4表示與上述所定義的相同的含義)的使用量優選相對於式(B)或式(B』)所表示的化合物為0.7~2.0當量,更優選為0.8~1.5當量。使用鹼時的鹼使用量優選相對於式(B)或式(B』)所表示的化合物為1.0~10.0當量,更優選為1.0~5.0當量。
反應溫度優選為20℃~100℃的範圍,更優選為40℃~80℃。反應時間優選為0.1小時~3天的範圍,更優選為1小時~2天的範圍。
特別優選的條件是,R4為氯原子,作為溶劑使用N,N-二甲基甲醯胺、N-甲基-2-吡咯烷酮、N,N-二甲基乙醯胺、N,N-二甲基甲醯胺與甲苯的混合溶劑、N,N-二甲基甲醯胺與二甲苯的混合溶劑、二甲苯與N-甲基-2-吡咯烷酮的混合溶劑或二甲苯與N,N-二甲基乙醯胺的混合溶劑,Ar-CH2-R4的使用量相對於式(B)或式(B』)所表示的化合物為0.8~1.5當量,反應溫度為40℃~80℃,反應時間為1小時~2天的條件,鹼使用1.0~5.0當量的碳酸鉀或三乙胺的條件。
從式(A)或式(A』)所表示的化合物經由式(B)或(B』)所表示的化合物得到式(I)或式(I』)所表示的化合物的一鍋製造
從式(A)或式(A』)所表示的化合物合成式(I)或式(I』)所表示的化合物時,可以在不分離式B或式(B』)所表示的化合物地進行之後的工序,從而得到式(I)或式(I』)所表示的化合物。
具體而言,將式(B)或式(B』)所表示的反應物直接、或減壓下除去過量的試劑後,添加Ar-CH2-R4(Ar、R4表示與上述相同的含義)和鹼,在上述條件下反應,得到式(I)或式(I』)所表示的化合物。
作為從式(A)或式(A』)所表示的化合物經由式(B)或(B』)所表示的化合物得到式(I)或式(I』)所表示的化合物的方法的優選例,是在鹼不存在下,使用芳香族烴系溶劑或非質子性極性溶劑、或其混合溶劑,使式(A)或式(A』)所表示的化合物與醯基化劑R1COR2反應而得到式(B)或式(B』)所表示的化合物後,加入芳香族烴系溶劑、或非質子性極性有機溶劑、或它們的混合溶劑、Ar-CH2-R4和鹼,直接或在減壓下餾去芳香族烴系溶劑的同時進行反應,得到式(I)或式(I』)所表示的化合物的方法。
在一鍋製造中從式(A)或式(A』)所表示的化合物製造式(B)或(B』)所表示的化合物
這裡所說的試劑的當量數,是相對於全部式(A)或式(A』)所表示的化合物的當量數。為了從式(A)或式(A』)所表示的化合物得到式(B)或式(B』)所表示的化合物,特別優選使用R2為CF3COO基、OEt基、羥基或氯原子的R1COR2、或CF3COR2。
R2為CF3COO基(例如,三氟乙酸酐)時,是作為溶劑使用甲苯,醯基化劑的使用量為1.0~1.5當量,反應溫度為-10℃~30℃,反應時間為0.5~4小時的條件,特別優選鹼不存在下,或在使用鹼的情況下使用碳酸鉀、其當量數使用1.0~3.0當量的條件。R2為OEt基(三氟乙酸乙酯)時,是作為溶劑使用N,N-二甲基甲醯胺、或N,N-二甲基甲醯胺與甲苯的混合溶劑,醯基化劑的使用量為1.5~5.0當量,反應溫度為40~80℃,反應時間為2小時~2天的條件,特別優選在鹼不存在下,或在使用鹼的情況下使用碳酸鉀或二甲基氨基吡啶、其當量數使用0.01~2.0當量的條件。
R2為羥基(例如,三氟乙酸)時的特別優選的條件是,作為溶劑使用甲苯、N,N-二甲基甲醯胺、二甲苯、N-甲基-2-吡咯烷酮、N,N-二甲基乙醯胺、N,N-二甲基甲醯胺與甲苯的混合溶劑、N,N-二甲基甲醯胺與二甲苯的混合溶劑、二甲苯與N-甲基-2-吡咯烷酮的混合溶劑或二甲苯與N,N-二甲基乙醯胺的混合溶劑,醯基化劑的使用量為1.0~3.0當量。使用亞硫醯氯、三氯氧磷、乙二醯氯時的特別優選的條件是,這些試劑使用0.3~3.0當量,在鹼不存在下,反應溫度為-10℃~40℃,反應時間為0.5小時~1天的條件。使用五氧化二磷、硫酸、多聚磷酸時的特別優選的條件是,這些試劑使用0.5~2.0當量,反應溫度為-10℃~160℃,反應時間為0.5小時~1天的條件。使用N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽時的特別優選的條件是,這些試劑使用0.5~3.0當量,反應溫度為-10℃~40℃,以三乙胺作為鹼使用0.5~3.0當量,反應時間為0.5~1天的條件。使用氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁、三氟化硼、對甲苯磺酸時的特別優選的條件是,這些試劑使用0.0001~0.5當量,在鹼不存在下,反應溫度為80℃~160℃,反應時間為2小時~2天的條件。
R2為氯原子(例如,三氟乙醯氯)時,是作為溶劑使用甲苯、N,N-二甲基甲醯胺、N-甲基-2-吡咯烷酮或它們的混合溶劑,醯基化劑的使用量為1.0~3.0當量,反應溫度為-30℃~30℃、反應時間為0.5小時~8小時的條件。特別優選在鹼不存在下,或使用鹼的情況下使用碳酸鉀、其當量數使用1.0~3.0當量的條件。
在一鍋製造中從式(B)或式(B』)所表示的化合物製造式(I)或(I』)所表示的化合物
用於從式(B)或式(B』)所表示的化合物得到式(I)或式(I』)所表示的化合物的特別優選的條件是,R4為氯原子,作為溶劑使用N,N-二甲基甲醯胺、N-甲基-2-吡咯烷酮、N,N-二甲基乙醯胺、N,N-二甲基甲醯胺與甲苯的混合溶劑、N,N-二甲基甲醯胺與二甲苯的混合溶劑、二甲苯與N-甲基-2-吡咯烷酮的混合溶劑或二甲苯與N,N-二甲基乙醯胺的混合溶劑,Ar-CH2-R4的使用量相對於式(B)或式(B』)所表示的化合物為0.8~1.5當量,反應溫度為40℃~80℃,反應時間為1小時~2天的條件,是鹼使用1.0~5.0當量的碳酸鉀或三乙胺的條件。
從式(Ba)所表示的化合物製造式(Ia)所表示的化合物的工序
作為從式(Ba)所表示的化合物得到式(Ia)所表示的化合物的方法,可以在無溶劑條件下或在不影響反應的溶劑中,使式(Ba)所表示的化合物在鹼存在下或不存在下,與醯基化劑R1aCOR2a(R1a、R2a表示與上述所定義的相同的含義)反應,從而得到。這裡所說的試劑的當量數,是相對於全部式(Ba)所表示的化合物的當量數。
作為可用的溶劑,可列舉甲苯、二甲苯、乙苯等芳香族烴系溶劑、乙酸乙酯、乙酸丁酯等酯系溶劑、乙醚、異丙醚、四氫呋喃、二烷等醚系溶劑、N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、N-甲基-2-吡咯烷酮、乙腈等非質子性極性有機溶劑、二氯甲烷、氯仿等滷素系溶劑、環己烷等烴系溶劑、丙酮、甲乙酮等酮系溶劑、水、或它們的混合溶劑。
作為可用的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鈉、碳酸氫鉀、氫氧化鈉、氫氧化鎂、氫氧化鈣、氫氧化鋰、氫氧化鋇等無機鹼、1,8-二氮雜二環[5.4.0]十一碳-7-烯、1,5-二氮雜二環[4.3.0]壬-5-烯、三乙胺、二異丙基乙基胺、吡啶、甲基吡啶、二甲基氨基吡啶等有機鹼、乙醇鈉、甲醇鈉、叔丁醇鉀等醇化物。可以不使用鹼,但在鹼存在下進行時的使用量可使用0.01~20.0當量。
作為醯基化劑R1COR2,可列舉三氟乙酸酐、三氟乙酸、三氟乙酸乙酯、三氟乙醯氯、或混合酸酐。另外,這樣的醯基化劑可以單獨使用或2種以上混合使用。其中,優選可使用三氟乙酸酐、三氟乙酸、三氟乙酸乙酯、或三氟乙醯氯。另外,R2表示羥基時,可以通過將N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽、1,1』-羰基二咪唑、二吡啶基二硫、二咪唑基二硫、1,3,5-三氯苯甲醯氯、1,3,5-三氯苯甲酸酐、PyBop(註冊商標)、PyBrop(註冊商標)、五氧化二磷、硫酸、多聚磷酸、亞硫醯氯、三氯氧磷、乙二醯氯、氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁、三氟化硼、對甲苯磺酸等試劑同時使用而進行。醯基化劑的使用量優選為0.5~10.0當量。
反應溫度優選為-80℃~200℃的範圍。反應時間優選為0.1小時~7天的範圍。
作為優選的方式,
(1)在R2表示三氟乙醯氧基時,具體地在作為醯基化劑使用三氟乙酸酐時,作為優選的溶劑,可列舉乙酸乙酯、乙酸丁酯等酯系溶劑、二氯甲烷、氯仿等滷素系溶劑、甲苯、二甲苯、乙苯等芳香族烴系溶劑,更優選為甲苯。反應優選在鹼不存在下進行,但在使用鹼的情況下,作為優選的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鉀、三乙胺、吡啶等,更優選為碳酸鉀。醯基化劑的使用量優選為1.0~5.0當量,更優選為1.0~1.5當量。使用鹼時的鹼使用量優選為1.0~4.5當量,更優選為1.0~3.0當量。反應溫度優選為-20℃~50℃的範圍,更優選為-10℃~30℃。反應時間優選為0.1小時~7天的範圍,更優選為0.5小時~4小時的範圍。
特別優選的條件是,作為醯基化劑使用三氟乙酸酐,作為溶劑使用甲苯,醯基化劑的使用量為1.0~1.5當量,反應溫度為-10℃~30℃,反應時間為0.5~4小時的條件。在鹼不存在下,或在使用鹼的情況下使用碳酸鉀、其當量數使用1.0~3.0當量的條件。
(2)在R2表示可被滷原子取代的C1-6烷氧基、或苯基可被滷原子、甲基、氰基、硝基、甲氧基取代的苄氧基時,具體在使用三氟乙酸乙酯、三氟乙酸甲酯、三氟乙酸丙酯、特別優選為三氟乙酸乙酯等時,作為優選的溶劑,是N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、N-甲基-2-吡咯烷酮、乙腈等非質子性極性有機溶劑、乙醚、異丙醚、四氫呋喃、二烷等醚系溶劑、及這些的溶劑與甲苯、二甲苯、乙苯等芳香族烴系溶劑的混合溶劑,更優選為N,N-二甲基甲醯胺、或N,N-二甲基甲醯胺與甲苯的混合溶劑。反應優選在鹼不存在下進行,但在使用鹼的情況下,作為優選的鹼,可列舉碳酸鉀、三乙胺、二甲基氨基吡啶等,更優選為碳酸鉀、二甲基氨基吡啶。醯基化劑的使用量優選為1.0~5.0當量,更優選為1.5~5.0當量。使用鹼時的鹼使用量優選為0.01~3.0當量,更優選為0.01~2.0當量。反應溫度優選為20℃~100℃的範圍,更優選為40℃~80℃。反應時間優選為0.1小時~7天的範圍,更優選為1小時~2天的範圍。
特別優選的條件是,作為醯基化劑使用三氟乙酸乙酯,作為溶劑使用N,N-二甲基甲醯胺、或N,N-二甲基甲醯胺與甲苯的混合溶劑,醯基化劑的使用量為1.0~5.0當量、反應溫度為40℃~80℃,反應時間為2小時~2天的條件。是在鹼不存在下,或在使用鹼的情況下使用碳酸鉀或二甲基氨基吡啶、其當量數使用0.01~2.0當量的條件。
(3)在R2表示可被滷原子取代的C1-6烷基羰氧基(但是,除了三氟乙醯氧基以外)、苯基可被滷原子、甲基、氰基、硝基、甲氧基取代的苯基羰氧基時,具體可列舉三甲基乙醯基。反應溫度優選為-20℃~50℃的範圍,更優選為-10℃~30℃。反應時間優選為0.1小時~7天的範圍,更優選為0.5小時~4小時的範圍。
(4)在R2表示羥基時,作為具體的醯基化劑可列舉三氟乙酸、二氟氯乙酸、三氯乙酸、二氟乙酸、二氯乙酸、二溴乙酸、氯乙酸、二氟丙酸、二氯丙酸、2,2,2-三氟丙酸、五氟丙酸、二氟環丙烷甲酸等,優選為三氟乙酸、三氯乙酸、二氯乙酸、二氟乙酸、二氟氯乙酸、氯乙酸、五氟丙酸,更優選為三氟乙酸、二氟乙酸、二氟氯乙酸、五氟丙酸,特別優選為三氟乙酸。使用三氟乙酸時,作為優選的溶劑,可列舉甲苯、二甲苯、乙苯等芳香族烴系溶劑及N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、N-甲基-2-吡咯烷酮、乙腈等非質子性極性有機溶劑,更優選為甲苯、二甲苯、N,N-二甲基甲醯胺、N-甲基-2-吡咯烷酮、甲苯與N,N-二甲基甲醯胺的混合溶劑、二甲苯與N,N-二甲基甲醯胺的混合溶劑或二甲苯與N-甲基-2-吡咯烷酮的混合溶劑。作為同時使用的試劑,可列舉N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽、五氧化二磷、硫酸、多聚磷酸、亞硫醯氯、三氯氧磷、乙二醯氯等,這些試劑優選使用0.2~5.0當量。另外,在作為同時使用的試劑使用氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁、三氟化硼、對甲苯磺酸等時,優選使用0.0001~1.0當量。反應在使用五氧化二磷、硫酸、多聚磷酸、亞硫醯氯、三氯氧磷、乙二醯氯、氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁、三氟化硼、對甲苯磺酸時,優選在鹼不存在下進行,在使用N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽時,優選在鹼存在下進行。使用鹼時作為優選的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鉀、三乙胺、吡啶、二甲基氨基吡啶等,更優選為三乙胺。醯基化劑的使用量優選為1.0~5.0當量,更優選為1.0~3.0當量。使用亞硫醯氯、三氯氧磷、乙二醯氯時,這些試劑使用0.2~5.0當量,反應溫度優選為-30℃~80℃的範圍,更優選為-10℃~40℃。使用五氧化二磷、硫酸、多聚磷酸時,這些試劑使用0.2~5.0當量,反應溫度優選為-30℃~200℃的範圍,更優選為-10℃~160℃。使用N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽時,優選這些試劑使用0.2~5.0當量,反應溫度優選為-30℃~80℃的範圍,更優選為-10℃~40℃,以三乙胺作為鹼使用0.2~5.0當量的條件。使用氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁、三氟化硼、對甲苯磺酸時,這些試劑使用0.0001~1.0當量,反應溫度優選為20℃~200℃的範圍,更優選為80℃~160℃。反應時間優選為0.1小時~7天的範圍,更優選為0.5小時~2天的範圍。
特別優選的條件是,作為醯基化劑使用三氟乙酸,作為溶劑使用甲苯、N,N-二甲基甲醯胺、二甲苯、N-甲基-2-吡咯烷酮、N,N-二甲基乙醯胺、N,N-二甲基甲醯胺與甲苯的混合溶劑、二甲苯與N,N-二甲基甲醯胺的混合溶劑、二甲苯與N-甲基-2-吡咯烷酮的混合溶劑或二甲苯與N,N-二甲基乙醯胺的混合溶劑,醯基化劑的使用量為1.0~3.0當量。使用亞硫醯氯、三氯氧磷、乙二醯氯時的特別優選的條件是,這些試劑使用0.3~3.0當量,在鹼不存在下,反應溫度為-10℃~40℃,反應時間為0.5小時~1天的條件。使用五氧化二磷、硫酸、多聚磷酸時的特別優選的條件是,這些試劑使用0.2~2.0當量,反應溫度為-10℃~160℃、反應時間為0.5小時~1天的條件。使用N,N』-二環己基碳二亞胺、1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽時的特別優選的條件是,這些試劑使用0.5~3.0當量,反應溫度為-10℃~40℃,以三乙胺作為鹼使用0.5~3.0當量,反應時間為0.5~1天的條件。使用氯化鋅、氯化銅、氯化鎂、氯化鈷、氯化鎳、氯化亞鐵、氯化鋁、硫酸鐵、硫酸鋁、三氟化硼、對甲苯磺酸時的特別優選的條件是,這些試劑使用0.0001~0.5當量,在鹼不存在下,反應溫度為80℃~160℃,反應時間為2小時~2天的條件。
(5)在R2表示滷原子時,具體地在使用三氟乙醯氯、三氟乙醯溴,優選為三氟乙醯氯時,作為優選的溶劑,可列舉甲苯、二甲苯、乙苯等芳香族烴系溶劑、二氯甲烷、氯仿等滷素系溶劑、及N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、N-甲基-2-吡咯烷酮、乙腈等非質子性極性有機溶劑,更優選為甲苯、N,N-二甲基甲醯胺、N-甲基-2-吡咯烷酮、或它們的混合溶劑。反應優選在鹼不存在下進行,但在使用鹼的情況下,作為優選的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鉀、三乙胺、吡啶等,更優選為碳酸鉀。醯基化劑的使用量優選為1.0~5.0當量,更優選為1.0~3.0當量。使用鹼時的鹼使用量優選為1.0~5.0當量,更優選為1.0~3.0當量。反應溫度優選為-80℃~40℃的範圍,更優選為-30℃~30℃。反應時間優選為0.1小時~7天的範圍,更優選為0.5小時~8小時的範圍。
另外,R2表示氯原子時,還可以使用在與式(Aa)所表示的化合物反應的體系外同時使用三氟乙酸與亞硫醯氯、三氯氧磷、乙二醯氯等而預先生成的R1COCl。
特別優選的條件是,作為醯基化劑使用三氟乙醯氯,作為溶劑使用甲苯、N,N-二甲基甲醯胺、N-甲基-2-吡咯烷酮或它們的混合溶劑,醯基化劑的使用量為1.0~3.0當量,反應溫度為-30℃~30℃,反應時間為0.5小時~8小時的條件。特別優選在鹼不存在下,或在使用鹼的情況下使用碳酸鉀、其當量數使用1.0~3.0當量的條件。
式(Ba)所表示的化合物可以通過專利文獻3等所記載的方法獲得。即,作為從式(Aa)所表示的化合物製造式(Ba)所表示的化合物的方法,可以在無溶劑條件下或在不影響反應的溶劑中,使式(Aa)所表示的化合物在鹼存在下或不存在下與式(Ca)所表示的化合物(X、R3、R4表示與上述所定義的相同的含義)所表示的化合物反應,從而得到。
作為可用的溶劑,可列舉乙醚、異丙醚、四氫呋喃、二烷等醚系溶劑、N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、乙腈、N-甲基-2-吡咯烷酮、N-甲基-2-哌嗪酮、N,N-二甲基-2-咪唑烷酮等非質子性極性有機溶劑、二氯甲烷、氯仿等滷素系溶劑、甲苯、二甲苯、乙苯等芳香族烴系溶劑、及它們的混合溶劑,優選列舉非質子性極性有機溶劑。更優選為N,N-二甲基甲醯胺、N,N-二甲基乙醯胺、甲苯。
即使不使用鹼也可實施反應,但作為使用鹼時的可用的鹼,可列舉碳酸鈉、碳酸鉀、碳酸氫鈉、碳酸氫鉀、氫氧化鈉、氫氧化鉀、氫氧化鎂、氫氧化鈣、氫氧化鋰、氫氧化鋇等無機鹼、1,8-二氮雜二環[5.4.0]十一碳-7-烯、1,5-二氮雜二環[4.3.0]壬-5-烯、三乙胺、二異丙基乙基胺、吡啶、二甲基吡啶、N,N-二甲基苯胺、N,N-二乙基苯胺、二甲基氨基吡啶等有機鹼,優選列舉碳酸鉀、三乙胺、吡啶等,更優選列舉三乙胺、碳酸鉀。
使用鹼時的鹼使用量優選相對於式(Aa)所表示的化合物為1.0~3.0當量,更優選為1.1~2.5當量。反應溫度優選為-20℃~150℃的範圍,更優選為-10℃~100℃。
反應時間優選為0.1小時~7天的範圍,更優選為1小時~2天。
另外,作為得到式(Ba)所表示的化合物其它方法,可列舉將所述式(Ib)所表示的化合物水解來製造式(Ba)所表示的化合物的方法(式中,R1、R3、X表示與所述的定義相同的含義)。
作為可用的溶劑,可列舉乙醚、異丙醚、四氫呋喃、二烷等醚系溶劑、N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、乙腈、N-甲基-2-吡咯烷酮、N-甲基-2-哌嗪酮、N,N-二甲基-2-咪唑烷酮等非質子性極性有機溶劑、二氯甲烷、氯仿等滷素系溶劑、甲苯、二甲苯、乙苯等芳香族烴系溶劑、甲醇、乙醇等醇系溶劑、水、及它們的混合溶劑,優選為芳香族烴系溶劑、或非質子性極性有機溶劑、或醇系溶劑與水的混合溶劑,更優選為N,N-二甲基甲醯胺與水、甲醇與水、或甲苯與水的混合溶劑。作為酸,可使用鹽酸、硫酸、磷酸、硝酸等無機酸,作為鹼,可使用碳酸鈉、碳酸鉀、碳酸氫鈉、碳酸氫鉀、氫氧化鈉、氫氧化鎂、氫氧化鈣、氫氧化鋰、氫氧化鋇等無機鹼。反應溫度優選為-20℃~150℃的範圍,更優選為70℃~100℃。反應時間優選為0.1小時~7天的範圍,更優選為1小時~8小時。
從式(Aa)所表示的化合物經由式(Ba)所表示的化合物合成式(Ia)所表示的化合物時,可以不分離式(Ba)所表示的化合物地進行之後的工序,得到式(Ia)所表示的化合物。
從式(A)或式(Aa)所表示的化合物合成式(I)或式(Ia)所表示的化合物時,可以使醯基化劑、溶劑、Ar-CH2-R4、鹼一次反應,而得到式(I)或式(Ia)所表示的化合物。
作為從式(A)或式(Aa)所表示的化合物使醯基化劑、溶劑、Ar-CH2-R4、鹼一次反應而得到式(I)或式(Ia)所表示的化合物時的優選例,是將式(A)或式(Aa)所表示的化合物,使用甲苯、二甲苯、乙苯等芳香族烴系溶劑、N,N-二甲基甲醯胺、二甲基亞碸、N,N-二甲基乙醯胺、乙腈、N-甲基-2-吡咯烷酮等非質子性溶劑或其混合溶劑,使用相對於式(A)或(Aa)所表示的化合物為1.0~5.0當量的R2表示可被滷原子取代的C1-6烷氧基的醯基化劑、具體為三氟乙酸乙酯、三氟乙酸甲酯、三氟乙酸丙酯等,作為鹼使用相對於式(A)或(Aa)所表示的化合物為1.0~10.0當量的碳酸鈉、碳酸鉀、碳酸氫鈉、碳酸氫鉀、氫氧化鈉、氫氧化鎂、氫氧化鈣、氫氧化鋰、氫氧化鋇等無機鹼、1,8-二氮雜二環[5.4.0]十一碳-7-烯、1,5-二氮雜二環[4.3.0]壬-5-烯、三乙胺、二異丙基乙基胺、吡啶、甲基吡啶、二甲基氨基吡啶等有機鹼、乙醇鈉、甲醇鈉、叔丁醇鉀等醇化物,加入相對於式(A)或(Aa)所表示的化合物為0.8~1.5當量的Ar-CH2-R4,在20℃~100℃下反應2小時~3天,得到式(I)或式(Ia)所表示的化合物的方法。
特別優選為使用甲苯、N,N-二甲基甲醯胺、或甲苯與N,N-二甲基甲醯胺的混合溶劑作為溶劑,使用三氟乙酸乙酯作為醯基化劑,Ar-CH2-R4的R4為氯原子,作為鹼使用碳酸鉀,相對於式(I)或式(Ia)所表示的化合物,醯基化劑的當量數優選為1.0~5.0當量,更優選為1.5~5.0當量,Ar-CH2-R4的當量數為0.8~1.5當量,鹼的當量數為1.0~5.0當量,反應溫度為40℃~80℃,反應時間為4小時~2天的條件。
從粗生成物純化分離式(I)所表示的化合物及式(Ia)所表示的化合物的方法
式(I)所表示的化合物及式(Ia)所表示的化合物可以通過單獨或組合通常使用的結晶化法、溶劑提取法、柱層析等進行純化分離。溶劑提取法所使用的溶劑只要是不與水混合的溶劑就不特別選擇,具體可列舉乙酸乙酯、乙酸丁酯、甲苯、乙苯、乙醚、異丙醚、二氯甲烷、氯仿等。結晶化法中使用的溶劑,可列舉水、己烷、甲苯、丙酮、N,N-二甲基甲醯胺、甲醇、2-丙醇、二氯甲烷、氯仿、乙酸乙酯、乙醚、二甲苯、N-甲基-2-吡咯烷酮、N,N-二甲基乙醯胺等及它們的混合溶劑。
式(I)所表示的化合物及式(Ia)所表示的化合物的優選的純化分離方法為結晶化法,作為結晶化溶劑優選單獨或組合使用丙酮、甲苯、水、N,N-二甲基甲醯胺、甲醇、二甲苯、N-甲基-2-吡咯烷酮、N,N-二甲基乙醯胺,更優選為水、甲醇、N,N-二甲基甲醯胺、N-甲基-2-吡咯烷酮、N,N-二甲基乙醯胺的組合。
實施例
本發明的具體例在以下顯示,但是本發明並不限於這些。
合成例1:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺(化合物1)的合成
(1)將25g(270mmol)2-氨基吡啶溶解於200ml無水二氯甲烷中,加入41ml(30g,300mmol)三乙胺並冷卻至0℃。在其中經過15分鐘滴加38ml(57g,270mmol)三氟乙酸酐,在室溫攪拌2小時。反應結束後,將反應液注入約100ml的冰水中,攪拌10分鐘。移到分液漏鬥進行分液,將有機層以150ml水洗滌2次,並以150ml的1%HCL水溶液洗滌2次後,以無水硫酸鎂進行乾燥,減壓濃縮而得到36g的2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺(收率71%)。
1H-NMR(CDCl3,δ,ppm):7.20(1H,m),7.83(1H,m),8.20(1H,d),8.35(1H,d),10.07(1H,brs)
13C-NMR(CDCl3,δ,ppm):115.3,115.5(q),121.6,139.1,147.9,149.5,155.3(q)MS:m/z=191(M+H)。
(2)將20g(126mmol)2-氯-5-氯甲基吡啶溶解於200ml無水乙腈,加入24g(126mmol)上述的方法所得到的2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺、21g(151mmol)碳酸鉀,加熱回流6小時後,在室溫攪拌10小時。反應結束後,過濾反應液,減壓濃縮濾液。在其中加入乙醚而結晶化,濾取所得到的晶體,以乙醚與水充分洗滌。所得到的晶體於60℃下減壓乾燥1小時,得到目的物。產量26g(收率66%)。
1H-NMR(CDCl3,δ,ppm):5.57(2H,s),6.92(1H,td),7.31(1H,d),7.80(1H,td),7.87(1H,dd),7.99(1H,dd),8.48(2H,m)
13C-NMR(CDCl3,δ,ppm):53.8,115.5,117.2(q),122.1,124.7,130.0,139.2,140.0,142.5,149.7,151.8,158.9,163.5(q)
MS:m/z=316(M+H)。
(3)粉末X射線晶體分析
在粉末X射線衍射中,用以下的條件進行測定。
裝置名:RINT-2200(株式會社リガク)
X射線:Cu-Kα(40kV、20mA)
掃描範圍:4~40°取樣寬度:0.02°掃描速度:1°/分鐘
結果如下(圖1)。
衍射角(2θ)8.7°、14.2°、17.5°、18.3°、19.8°、22.4°、30.9°、35.3°。
(4)差示掃描量熱分析(DSC)
在差示掃描量熱分析中,在以下的條件下測定。
裝置名:DSC-60
樣品盒:鋁
溫度範圍:50℃~250℃(升溫:10℃/分鐘)
結果在圖2中顯示。
(5)另外,通過以下的(i)~(iv)所記載的方法(第二~五的製法)進行再晶體而得到同等的晶體。對這些晶體,以與上述同樣的測定條件,進行粉末X射線晶體分析及差示掃描量熱分析。
(i)第二製法
在化合物1(700mg)中加入約25ml己烷、約25ml乙酸乙酯,在熱水浴中加熱至65℃使其完全溶解。將其緩緩恢復到室溫並放置一夜。濾出所析出的晶體,以少量的己烷:乙酸乙酯=95:5的溶液洗滌晶體。將其放入乾燥器在減壓下乾燥2小時,得到白色晶體349mg。
粉末X射線晶體分析的結果如下(圖3)。
衍射角(2θ)8.5°、14.0°、17.3°、18.1°、19.6°、22.2°、30.8°、35.2°
差示掃描量熱分析的結果在圖4中顯示。
(ii)第三製法
在化合物1(1.0g)中加入28ml 2-丙醇,在熱水浴中加熱至65℃使其完全溶解。使其緩緩恢復到室溫並放置一夜。濾出所析出的晶體,以少量的2-丙醇洗滌後,置入乾燥器中減壓下乾燥2小時,得到白色晶體695mg。
差示掃描量熱分析的結果在圖5中顯示。
(iii)第四製法
在化合物1(700mg)中加入約30ml甲苯,在熱水浴中加熱至65℃使其完全溶解。將其緩緩恢復到室溫並放置一夜。濾出所析出的晶體,以少量的甲苯洗滌後,置入乾燥器中減壓下乾燥2小時,得到白色晶體440mg。
粉末X射線晶體分析的結果如下(圖6)。
衍射角(2θ)8.6°、14.2°、17.5°、18.3°、19.7°、22.3°、30.9°、35.3°
差示掃描量熱分析的結果在圖7中顯示。
(iv)第五製法
在化合物1(50mg)中加入甲醇約2ml、水約2ml,在熱水浴中加熱至65℃使其溶解。將其恢復到室溫並放置一夜。濾出所析出的晶體,得到白色晶體16mg。
差示掃描量熱分析的結果在圖8中顯示。
表1顯示依照合成例1的方法所製造的式(I)所表示的有害生物防除劑的具體化合物例及其物性值。
表1
合成例2:2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺的合成
將1.0g(10.6mmol)2-氨基吡啶溶解於10ml乙酸乙酯中,加入1.78ml(12.7mmol)三乙胺,冰冷下添加1.62ml(11.7mmol)三氟乙酸酐。然後在室溫攪拌2小時後,加入10ml乙酸乙酯與10ml水並攪拌後進行分液。將乙酸乙酯層進一步以10ml水洗滌二次後,以無水硫酸鎂乾燥,減壓濃縮,得到2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺1.56g(77.2%)。
合成例3:2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺的合成
將4.7g(50mmol)2-氨基吡啶溶解於25ml N,N-二甲基甲醯胺中,添加35.5g(250mmol)三氟乙酸乙酯。然後在55~60℃攪拌15小時後,加入100ml乙酸乙酯和100ml水並攪拌後進行分液。將乙酸乙酯層進一步以100ml水及100mL食鹽水洗滌後,以無水硫酸鎂乾燥,減壓濃縮,得到2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺9.05g(95.6%)。
1H-NMR(CDCl3,δ,ppm):7.20(1H,ddd),7.83(1H,td),8.20(1H,d),8.35(1H,d),10.07(1H,brs)。
合成例4:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將50.0g(0.53mol)2-氨基吡啶溶解於400ml甲苯後,在5℃冷卻下,將88.6ml(0.64mol)三氟乙酸酐以30分鐘滴加。滴加後在室溫攪拌30分鐘,減壓下將20ml甲苯蒸餾。在反應液中加入250ml二甲基甲醯胺,冰冷下緩緩加入88.2(0.64mol)g粉末碳酸鉀。然後,加入89.2g(0.557mol)2-氯-5-氯甲基吡啶,在減壓(50-60hPa)下,在40-45℃將甲苯慢慢蒸餾,加熱1小時。以60-70℃、35hPa進一步加熱蒸餾2.5小時後,加入5.0g(0.036mol)粉末碳酸鉀,以50-60℃、35hPa進一步1小時以除去水。將反應液添加在2L 50℃的水中,添加結束後,攪拌30分鐘。然後,過濾,以200ml、接著以500ml水進行漿洗滌。過濾後,以甲苯100ml進行壓洗,進一步以400ml甲苯進行漿洗滌。將所得到的晶體以60℃、真空泵減壓下進行乾燥一夜,得到作為目的的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺147.78g(88.1%)。取8.21g的所得到的標題化合物,溶解於100mL丙酮,在其中加入300mL水。在室溫攪拌並濾取所析出的晶體,將所得到的晶體以60℃、真空泵減壓下乾燥一夜,得到7.28g。所得到的晶體的粉末X射線晶體分析的結果如下(圖9)。
衍射角(2θ)8.8°、14.3°、17.6°、18.3°、19.9°、22.5°、31.0°、35.4°
合成例5:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將50.0g(0.53mol)2-氨基吡啶溶解於250ml甲苯後,在5℃冷卻下,將88.6ml(0.64mol)三氟乙酸酐以30分鐘滴加。滴加後在室溫攪拌30分鐘,減壓下蒸餾20ml甲苯。在反應液中加入250ml二甲基甲醯胺,在冰冷下,緩緩加入88.2g(0.64mol)粉末碳酸鉀。然後,加入87.0g(0.54mol)2-氯-5-氯甲基吡啶,在減壓(50-60hPa)下、以50-60℃緩緩蒸餾甲苯,進一步在35hPa下加熱。1小時後,加入5.0g(0.036mol)粉末碳酸鉀,以50-60℃、35hPa除去水。4小時後,將反應液添加在1.1L的50℃的水中,反應容器用150ml甲醇洗滌,並補加添加。添加結束後,在50℃加熱10分鐘,慢慢冷卻,在15-20℃攪拌30分鐘後,過濾,以150ml水、接著以150ml甲苯洗滌。漿所得到的晶體以60℃、真空泵減壓下乾燥11小時,得到作為目的的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺147.32g(87.8%)。
合成例6:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將1.0g(10.6mmol)2-氨基吡啶溶解於10ml甲苯中,在5℃冷卻後,加入1.18ml(15.9mmol)三氟乙酸,然後再加入0.99ml(10.6mmol)三氯氧磷,室溫下攪拌6.5小時。在反應液中加入5.0ml二甲基甲醯胺與5.87g(42.5mmol)粉末碳酸鉀及1.72g(10.6mmol)2-氯-5-氯甲基吡啶,在減壓下(60-35hPa)、以50-60℃蒸餾。2.5小時後,將反應液添加到100ml的水中,過濾晶體,以30ml水和15ml甲苯洗滌。所得到的晶體減壓下60℃乾燥,得到作為目的的化合物N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺2.09g(62.3%)。
合成例7:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將10.0g(0.106mol)2-氨基吡啶溶解於100ml甲苯中,在5℃冷卻後,加入11.8ml(0.159mol)三氟乙酸,然後再加入9.9ml(0.106mol)氯氧化磷,室溫下攪拌一夜,在減壓下,將20ml甲苯餾去。在冰冷下,在反應液中加入50ml二甲基甲醯胺與35.28g(0.256mol)粉末碳酸鉀、及17.22g(0.106mol)2-氯-5-氯甲基吡啶,在減壓下(60-35hPa)、以50-60℃蒸餾。2小時後,進一步加入25ml二甲基甲醯胺、20ml甲苯與7.35g(0.053mol)粉末碳酸鉀,在減壓下(60-35hPa)、以50-60℃蒸餾2小時。在反應液中加入60ml甲醇與50ml水,在一併洗滌容器的同時將反應液添加到300ml的水中,30分鐘後,過濾晶體,以70ml水與40ml甲苯洗滌。所得到的晶體在減壓下60℃乾燥,得到作為目的的化合物25.75g(76.9%)。
合成例8:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將10.0g(0.106mol)2-氨基吡啶溶解於100ml甲苯,在5℃冷卻後,加入11.8ml(0.159mol)三氟乙酸,然後再加入7.7ml(0.106mol)亞硫醯氯,在室溫下攪拌一夜,減壓下將20ml甲苯餾去。在冰冷下,在反應液中加入50ml二甲基甲醯胺、35.28g(0.256mol)粉末碳酸鉀與17.22g(0.106mol)2-氯-5-氯甲基吡啶,在減壓下(36hPa)、以50-60℃蒸餾一小時。在反應液中加入60ml甲醇與50ml水,在一併洗滌容器的同時將反應液添加到300ml的水中,30分鐘後,過濾晶體,以70ml水與40ml甲苯洗滌。所得到的晶體在減壓下60℃乾燥,得到作為目的的化合物22.31g(66.6%)。
合成例9:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將5.0g(0.053mol)2-氨基吡啶溶解於50ml甲苯後,在5℃冷卻下,將8.86ml(0.064mol)三氟乙酸酐以10分鐘滴加。滴加後於室溫攪拌30分鐘,在減壓下蒸餾10ml甲苯。在反應液中加入25ml二甲基甲醯胺,冰冷下,緩緩加入8.82g粉末碳酸鉀。然後,加入11.78g(0.053mol)2-氯-5-甲磺醯氧基甲基吡啶,在減壓(50-60hPa)下、以50-60℃慢慢蒸餾甲苯,進一步在35hPa進行加熱。30分鐘後,加入30ml二甲基甲醯胺、30ml甲苯與1.18g(0.0053mol)2-氯-5-甲磺醯氧基甲基吡啶,以50-60℃、55hPa進行減壓蒸餾。4小時後,將反應液添加到250ml水中,將反應容器用30ml甲醇與20ml水洗滌並補加添加。添加結束後,於室溫攪拌30分鐘後,過濾,以50ml水接著再以40ml甲苯洗滌。將所得到的晶體以80℃、真空泵減壓下乾燥11小時,得到作為目的的N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺11.63g(69.4%)。
合成例10:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將10.0g(0.106mol)2-氨基吡啶溶解於100ml甲苯,在5℃冷卻後,加入11.84ml(0.159mol)三氟乙酸,然後再加入5.94ml(0.064mol)三氯氧磷,於室溫下攪拌一夜,減壓下,餾去20ml甲苯。在冰冷下在反應液中加入50ml二甲基甲醯胺與22.03g(0.16mol)粉末碳酸鉀及17.56g(0.108mol)2-氯-5-氯甲基吡啶,以減壓下(60-35hPa)、以50-60℃蒸餾。1小時後,進一步加入20ml二甲基甲醯胺、20ml甲苯與4.41g(0.032mol)粉末碳酸鉀,在減壓下(60-35hPa)、以50-60℃蒸餾1.5小時。在反應液中加入30ml甲醇,然後添加到250ml的50℃的水中,然後再加入水50ml,在洗滌容器的同時添加,於室溫冷卻後攪拌30分鐘,過濾晶體,以50ml水與30ml甲苯洗滌。所得到的晶體在減壓下60℃進行乾燥,得到作為目的的化合物23.69g(70.6%)。
合成例11:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將10.0g(0.106mol)2-氨基吡啶溶解於100ml甲苯,在5℃冷卻後,加入11.8ml(0.159mol)三氟乙酸,然後再分次加入7.76ml(0.106mol)亞硫醯氯,於室溫下攪拌一夜,減壓下,餾去50ml甲苯。在反應液中加入50ml甲苯,冰冷下加入50ml二甲基甲醯胺、22.03g(0.16mol)粉末碳酸鉀與17.56g(0.108mol)2-氯-5-氯甲基吡啶,在減壓下(90-36hPa)、以60℃蒸餾1.5小時。在反應液中加入30ml甲醇與20ml水,與一併洗滌容器的同時將反應液添加到50℃的300mL的水中,於室溫30分鐘攪拌後,過濾晶體,以50ml水與30ml甲苯洗滌。所得到的晶體以減壓下60℃乾燥,得到作為目的的化合物21.45g(64.1%)。
合成例12:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將94g(1mol)2-氨基吡啶溶解於500mL二甲基甲醯胺,加入284g(2mol)三氟乙酸乙酯,於55-60℃攪拌24小時。在反應液中加入82.8g(0.6mol)粉末碳酸鉀與153.9g(0.95mol)2-氯-5-氯甲基吡啶及300mL甲苯,在減壓下(36hPa)、以50-60℃攪拌3小時。在反應液中添加200mL甲醇後,將反應液添加到2L的50℃的溫水中,放冷至室溫後,攪拌3小時。過濾晶體,以400mL水及450mL甲苯洗滌。所得到的晶體以減壓下45℃乾燥,得到作為目的的化合物228.9g(收率72.7%)。
合成例13:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將9.4g(0.1mol)2-氨基吡啶溶解於30mL二甲基甲醯胺及20mL甲苯的混合溶劑中,加入28.4g(0.2mol)三氟乙酸乙酯,於60-65℃攪拌8小時。在反應液中加入16.6g(0.12mol)粉末碳酸鉀與16.2g(0.1mol)2-氯-5-氯甲基吡啶,以60-65℃攪拌15小時。在反應液中添加15mL甲醇後,在120mL的50℃的溫水中添加反應液,放冷至室溫後,攪拌2小時。過濾晶體,以50mL水及100mL甲苯洗滌。所得到的晶體以減壓下45℃乾燥,得到作為目的的化合物25.6g(收率81.2%)。
合成例14:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
在13.68g(0.12mol)三氟乙酸中添加1.5mL二甲基甲醯胺,在65℃加溫的同時加入14.28g(0.12mol)亞硫醯氯。將由此產生的三氟乙醯氯鼓泡至將9.4g(0.1mol)2-氨基吡啶溶解於50mL N-甲基吡咯烷酮並於-10℃冷卻而得的溶液中,攪拌1小時。在該反應液中加入100mL甲苯、48.3g(0.35mol)粉末碳酸鉀與16.52g(0.102mol)2-氯-5-氯甲基吡啶,減壓下(36hPa)、以50-60℃蒸餾3小時。在反應液中加入20mL甲醇,在一併洗滌容器的同時添加到加溫至50℃的300mL的水中。以室溫攪拌1.5小時後,過濾晶體,以100mL水及150mL甲苯洗滌。將所得到的晶體以減壓下45℃乾燥,得到作為目的的化合物16.8g(收率53.3%)。
合成例15:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
在18.24g(0.16mol)三氟乙酸中添加8.76g(0.12mol)二甲基甲醯胺,在65℃加溫的同時加入12.26g(0.08mol)三氯氧磷。將由此產生的三氟乙醯氯鼓泡至將9.4g(0.1mol)2-氨基吡啶溶解於80mL N-甲基吡咯烷酮於-15℃冷卻而得的溶液中,攪拌2小時。在-10℃冷卻的同時,在該反應液中加入14.9g(0.22mol)粉末乙醇鈉,進行中和。在反應液中加入13.8g(0.1mol)粉末碳酸鉀、及16.2g(0.1mol)2-氯-5-氯甲基吡啶,減壓下(36hPa)、以50-60℃蒸餾2小時。在反應液中加入20mL甲醇,在一併洗滌容器的同時添加到加溫至50℃的400mL的水中。於室溫攪拌30分鐘後,過濾晶體,以100mL水及50mL甲苯洗滌。所得到的晶體以減壓下45℃乾燥,得到作為目的的化合物22.5g(收率71.4%)。
合成例16:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將3.00g(18.6mmol)2-氯-5-氯甲基吡啶溶解於20ml二甲基甲醯胺中,加入1.75g(18.6mmol)2-氨基吡啶在80℃攪拌8小時、在室溫攪拌5小時。反應結束後,減壓餾去二甲基甲醯胺,加入乙腈,結果析出固體,因此濾出,以乙腈徹底洗滌後進行乾燥,得到1-[(6-氯吡啶-3-基)甲基]吡啶-2(1H)-亞胺鹽酸鹽2.07g(收率44%)。
1H-NMR(DMSO-d6,δ,ppm):5.65(2H,s),6.96(1H,t),7.23(1H,m),7.57(1H,d),7.80(1H,m),7.91(1H,m),8.28(1H,m),8.49(1H,d)。
將50mg(0.20mmol)以上述的方法得到的1-[(6-氯吡啶-3-基)甲基]吡啶-2(1H)-亞胺鹽酸鹽懸浮於5ml無水二氯甲烷中,在冰冷下依次加入122mg(1.00mmol)二甲基氨基吡啶、50mg(0.24mmol)三氟乙酸酐,在室溫攪拌1小時。反應結束後,將反應液以二氯甲烷稀釋,以1%鹽酸洗滌後,以無水硫酸鎂乾燥。減壓餾去二氯甲烷而得到目的物。產量42mg(收率67%)。
合成例17:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
按照合成例16的方法合成後,將4.6g(0.02mol)中和而得到的1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞胺溶解於15mL N,N-二甲基甲醯胺中,加入5.7g(0.04mol)三氟乙酸乙酯。於56℃攪拌整夜後,加入60mL水,濾取所析出的晶體。將所得到的晶體在減壓下45℃下乾燥,得到作為目的的化合物5.85g(收率92.8%)。
合成例18:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
按照合成例16的方法合成後,將2.2g(0.01mol)中和而得到的1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞胺溶解於6mL N,N-二甲基甲醯胺中,冰冷下,加入828mg(0.006mol)碳酸鉀及2.52g(0.012mol)三氟乙酸酐。於室溫攪拌1小時後,加入30mL水,濾取所析出的晶體。將所得到的晶體以20mL水洗滌後,減壓下45℃下乾燥,得到作為目的的化合物2.38g(收率75.6%)。
合成例19:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
在4.56g(0.04mol)三氟乙酸中添加3mL N,N-二甲基甲醯胺,在60℃加溫的同時加入3.12g(0.02mol)三氯氧磷。將由此產生的三氟乙醯氯,鼓泡至將4.38g(0.02mol)按照合成例16的方法合成後、中和而得到的1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞胺溶解於25mL N-甲基-2-吡咯烷酮而得的溶液中,於-10℃使其反應45分鐘。加入125mL水,濾取所析出的晶體。將所得到的晶體在減壓下45℃下乾燥,得到作為目的的化合物2.58g(收率40.9%)。
合成例20:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
按照合成例16的方法合成後,將4.38g(0.02mol)中和而得到的1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞胺溶解於3mL N,N-二甲基甲醯胺中,加入2.7g(0.024mol)三氟乙酸及2.8g(0.02mol)五氧化二磷。於120℃攪拌3小時後,回到室溫,加入50mL水,濾取所析出的晶體。將所得到的晶體在減壓下45℃下乾燥,得到作為目的的化合物2.12g(收率33.7%)。
合成例21:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將9.4g(0.1mol)2-氨基吡啶溶解於50mL二甲基甲醯胺,加入28.8g(0.2mol)三氟乙酸乙酯與16.2g(0.1mol)2-氯-5-氯甲基吡啶、及13.8g(0.1mol)碳酸鉀,於55-60℃攪拌20小時。在反應液中進一步加入1.38g(0.1mol)粉末碳酸鉀與3.24(0.02mol)2-氯-5-氯甲基吡啶、及5.68g(0.04mol)三氟乙酸乙酯,於55-60℃攪拌6小時。在反應液中添加40mL甲醇然後,將反應液添加於300mL 50℃的溫水中,放冷至室溫後,攪拌1小時。過濾晶體,以100mL水及75mL甲苯洗滌。所得到的晶體在減壓下45℃乾燥,得到作為目的的化合物24.0g(收率76%)。
合成例22:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將9.4g(0.1mol)2-氨基吡啶溶解於30mL二甲基甲醯胺及20mL甲苯中,加入28.8g(0.2mol)三氟乙酸乙酯與16.2g(0.1mol)2-氯-5-氯甲基吡啶、及16.6g(0.12mol)碳酸鉀,於60-65℃攪拌18小時。添加15mL甲醇於反應液中後,添加反應液於120mL 50℃的溫水中,放冷至室溫後,攪拌1小時。過濾晶體,以50mL水及100mL甲苯洗滌。所得到的晶體在減壓下45℃下乾燥,得到作為目的的化合物23.9g(收率75.9%)。
合成例23:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將4.7g(0.05mol)2-氨基吡啶溶解於25mL N,N-二甲基甲醯胺與10mL甲苯的混合溶劑中,加入35.5g(0.25mol)三氟乙酸乙酯與9.72g(0.06mol)2-氯-5-氯甲基吡啶、及8.28g(0.06mol)粉末碳酸鉀,於65℃攪拌18小時。添加10mL甲醇於反應液後,添加反應液於150mL 50℃的溫水中,放冷至室溫後,攪拌1小時。過濾晶體,以50mL水及50mL甲苯洗滌。所得到的晶體在減壓下45℃下乾燥,得到作為目的的化合物13.78g(收率87.5%)。
合成例24:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將9.4g(0.1mol)2-氨基吡啶溶解於30mL二甲基甲醯胺及20mL甲苯中,加入14.2g(0.1mol)三氟乙酸乙酯於60-65℃攪拌7小時。接著加入16.2g(0.1mol)2-氯-5-氯甲基吡啶、及16.6g(0.12mol)碳酸鉀,於60-65℃攪拌18小時。添加15mL甲醇於反應液然後,添加反應液於150mL 50℃的溫水中,放冷至室溫。過濾晶體,以50mL水及75mL甲苯洗滌。所得到的晶體在減壓下60℃下乾燥,得到作為目的的化合物20.6g(收率65.4%)。
合成例25:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將9.4g(0.1mol)2-氨基吡啶溶解於30mL二甲基甲醯胺及20mL甲苯中,加入7.1g(0.05mol)三氟乙酸乙酯於60-65℃攪拌7.5小時。經減壓濃縮(90hPa、40℃)後,進行冰冷,加入20mL甲苯及10.5g(0.05mol)三氟乙酸酐,於室溫攪拌1小時。接著,加入16.2g(0.1mol)2-氯-5-氯甲基吡啶、20mL二甲基甲醯胺、及16.6g(0.12mol)碳酸鉀,於110hPa的減壓下、於60-65℃攪拌4小時。經減壓濃縮(90hPa、50℃)後,添加25mL甲醇於反應液中,將其加入250mL 50℃的溫水中,放冷至室溫並同時攪拌。過濾晶體,以90mL水及90mL甲苯洗滌。所得到的晶體在減壓下60℃下乾燥,得到作為目的的化合物19.8g(收率62.9%)。
合成例26:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將9.4g(0.1mol)2-氨基吡啶溶解於30mL二甲基甲醯胺及20mL甲苯中,加入21.3g(0.15mol)三氟乙酸乙酯於60-65℃攪拌7.5小時。經減壓濃縮(90hPa、40℃)後,進行冰冷,加入20mL甲苯及10.5g(0.05mol)三氟乙酸酐,於室溫攪拌1小時。接著,加入16.2g(0.1mol)2-氯-5-氯甲基吡啶、20mL二甲基甲醯胺、及16.6g(0.12mol)碳酸鉀,於110hPa的減壓下,於60-65℃攪拌4小時。經減壓濃縮(90hPa、50℃)後,添加25mL甲醇於反應液中,將其加入250mL 50℃的溫水中,放冷至室溫同時攪拌。過濾晶體,以90mL水及90mL甲苯洗滌。所得到的晶體在減壓下60℃下乾燥,得到作為目的的化合物22.68g(收率72.0%)。
合成例27:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺的合成
將2.35g(0.025mol)2-氨基吡啶懸浮於40mL二甲苯中,加入2.85g(0.025mol)三氟乙酸及135mg氯化亞鐵6水合物,在加入迪安-斯達克回流裝置除去生成的水的同時,於150℃攪拌16小時。冷卻至60℃後,加入4.05g(0.025mol)2-氯-5-氯甲基吡啶、16mL二甲基甲醯胺、及2.42g(0.0175mol)碳酸鉀,於60-110hPa的減壓下,於60-65℃攪拌3小時。添加10mL甲醇於反應液中,將其加入80mL的50℃的溫水中,放冷至室溫同時攪拌。過濾晶體,以20mL水及20mL甲苯洗滌。所得到的晶體在減壓下60℃下乾燥,得到作為目的的化合物6.32g(收率80.3%)。
合成例28:N-[1-((6-氯-5-氟吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺(化合物2)的合成
將4.00g(27.6mmol)2-氯-3-氟-5-甲基吡啶溶於80ml四氯化碳中,加入7.37g(41.4mmol)N-溴琥珀醯亞胺、20mg過氧化苯甲醯加熱回流一夜。反應結束後,使反應液回到室溫進行減壓濃縮,用矽膠柱層析(己烷:乙酸乙酯=19:1)進行純化得到5-(溴甲基)-2-氯-3-氟吡啶3.06g(收率51%)。
1H-NMR(CDCl3,δ,ppm):4.45(2H,s),7.54(1H,dd),8.23(1H,s)。
將50mg(0.22mmol)上述的方法所得到的5-(溴甲基)-2-氯-3-氟吡啶溶於5ml無水乙腈,依次加入42mg(0.22mmol)所述的方法所得到的2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺、36mg(0.26mmol)碳酸鉀,並加熱回流7小時。反應結束後,使反應液回到室溫過濾不溶物,減壓濃縮濾液。在其中加入乙醚,結果析出固體,因而進行濾取,以乙醚洗滌後以乾燥器減壓乾燥得到目的物。產量29mg(收率40%)。
1H-NMR(CDCl3,δ,ppm):5.54(2H,s),6.89(1H,td),7.76(1H,dd),7.80(1H,td),7.85(1H,d),8.29(1H,d),8.57(1H,d)
MS:m/z=334(M+H)。
合成例29:N-[1-((6-溴吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺(化合物3)的合成
將500mg(2.92mmol)2-溴-5-甲基吡啶溶解於15ml四氯化碳中,加入623mg(3.50mmol)N-溴琥珀醯亞胺、10mg過氧化苯甲醯並加熱回流19小時。反應結束後,使反應液回到室溫進行減壓濃縮,以矽膠柱層析(己烷:乙酸乙酯=19:1)進行純化得到2-溴-5-溴甲基吡啶143mg(收率20%)。
1H-NMR(CDCl3,δ,ppm):4.42(2H,s),7.47(1H,d),7.59(1H,dd),8.38(1H,d)。
將70mg(0.28mmol)以上述的方法所得到的2-溴-5-溴甲基吡啶溶於10ml無水乙腈中,依次加入54mg(0.28mmol)所述的方法所合成的2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺、46mg(0.34mmol)碳酸鉀,並加熱回流6小時。反應結束後,使反應液回到室溫過濾不溶物,減壓濃縮濾液。在其中加入乙醚,結果析出固體,因而進行濾取,以乙醚洗滌後以乾燥器進行減壓乾燥得到目的物。產量81mg(收率82%)。
1H-NMR(CDCl3,δ,ppm):5.52(2H,s),6.88(1H,t),7.48(1H,d),7.78(2H,m),7.84(1H,d),8.44(1H,d),8.53(1H,d)
MS:m/z=360(M+H)。
合成例30:2-氯-N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2-二氟乙醯胺(化合物4)的合成
將200mg(2.13mmol)2-氨基吡啶溶解於5ml二氯甲烷中,依次加入491mg(2.55mmol)EDC-HCl、311mg(2.55mmol)二甲基氨基吡啶、187μl(2.23mmol、290mg)氯二氟乙酸,並攪拌一夜。反應結束後,將反應液以二氯甲烷稀釋,以水、1%鹽酸洗滌後,以無水硫酸鎂乾燥得到2-氯-2,2-二氟-N-(吡啶-2(1H)-亞基)乙醯胺105mg(收率24%)。
1H-NMR(CDCl3,δ,ppm):7.19(1H,dd),7.82(1H,m),8.18(1H,d),8.36(1H,d),9.35(1H,brs)。
在68mg(0.33mmol)所述的方法所合成的2-氯-2,2-二氟-N-(吡啶-2(1H)-亞基)乙醯胺中,加入溶解於6ml無水乙腈的53mg(0.33mmol)2-氯-5-氯甲基吡啶,接著加入50mg(0.36mmol)碳酸鉀加熱回流1小時。反應結束後,使反應液回到室溫然後進行減壓濃縮。在其中加入乙醚,結果析出固體,因而進行濾取,進行乾燥而得到目的物。產量49mg(收率45%)。
1H-NMR(CDCl3,δ,ppm):5.56(2H,s),6.92(1H,t),7.33(1H,d),7.82(1H,m),7.91(1H,dd),8.02(1H,d),8.45(1H,d),8.48(1H,d)
13C-NMR(CDCl3,δ,ppm):53.8,115.2,120.1(t),122.1,124.8,139.0,140.0,142.3,150.0,151.9,159.1,159.1,165.8(t)
MS:m/z=332(M+H)。
合成例31:2,2,2-三氯-N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]乙醯胺(化合物5)的合成
將70mg(0.27mmol)合成例16的方法所得到的1-[(6-氯吡啶-3-基)甲基]吡啶-2(1H)-亞胺鹽酸鹽懸浮於4ml無水二氯甲烷中,依次加入94μl(0.68mmol、68mg)三乙胺、33μg(0.27mmol、49mg)三氯乙醯氯,室溫攪拌1小時。反應結束後,加入水使反應停止,以二氯甲烷與水進行分液。將有機層以水洗滌1次、以1%鹽酸洗滌2次後,以無水硫酸鎂乾燥、減壓濃縮。在其中加入乙醚,結果析出固體,因而進行濾取,乾燥得到目的物。產量61mg(收率62%)。
1H-NMR(CDCl3,δ,ppm):5.59(2H,s),6.86(1H,t),7.32(1H,d),7.78(1H,td),7.91(2H,m),8.43(1H,d),8.50(1H,d)
MS:m/z=364(M+H)。
合成例32:N-[1-((2-氯嘧啶-5-基)甲基)吡啶-2(1H)-亞基]-2,2,2-三氟乙醯胺(化合物6)的合成
將1.04g(8.13mmol)2-氯-5-甲基嘧啶溶於30ml四氯化碳中,加入1.73g(9.75mmol)N-溴琥珀醯亞胺、20mg過氧化苯甲醯,加熱回流6小時。反應結束後,使反應液回到室溫進行減壓濃縮,以矽膠柱層析(己烷:乙酸乙酯=3:1)進行純化,得到5-溴甲基-2-氯嘧啶641mg(收率38%)。
1H-NMR(CDCl3,δ,ppm):4.42(2H,s),8.66(2H,s)。
將104mg(0.50mmol)上述的方法所得到的5-溴甲基-2-氯嘧啶溶解於6ml無水乙腈中,加入96mg(0.50mmol)所述的方法所得到的2,2,2-三氟-N-(吡啶-2(1H)-亞基)乙醯胺、76mg(0.55mmol)碳酸鉀,加熱回流1小時。反應結束後,使反應液回到室溫過濾不溶物並除去,減壓濃縮濾液。在其中加入乙醚,結果析出固體,因而進行濾取,以乙醚洗滌後放入乾燥器中進行減壓乾燥,得到目的物。產量92mg(收率58%)。
1H-NMR(CDCl3,δ,ppm):5.54(2H,s),6.98(1H,m),7.87(1H,m),8.18(1H,m),8.48(1H,m),8.83(2H,m)
13C-NMR(CDCl3,δ,ppm):0.0,115.6,117.1(q),122.1,127.5,139.2,142.9,158.8,160.3(2C),161.4,163.8(q)
MS:m/z=317(M+H)。
合成例33:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2,3,3,3-五氟丙醯胺(化合物7)的合成
將300mg(3.19mmol)2-氨基吡啶溶解於15ml無水二氯甲烷中,依次加入919mg(4.78mmol)EDC-HCl、583mg(4.78mmol)DMAP、397μl(628mg、3.83mmol)五氟丙酸,於室溫攪拌一夜。反應結束後,將反應液以二氯甲烷稀釋,以水洗滌1次,以1%鹽酸洗滌2次後,以無水硫酸鎂乾燥,減壓濃縮,得到2,2,3,3,3-五氟-N-(吡啶-2(1H)-亞基)丙醯胺85mg(收率11%)。
在77mg(0.32mmol)上述的方法所得到的2,2,3,3,3-五氟-N-(吡啶-2(1H)-亞基)丙醯胺中,加入溶解於8ml無水乙腈的52mg(0.32mmol)2-氯-5-氯甲基吡啶、49mg(0.35mmol)碳酸鉀,加熱回流11小時。反應結束後,使反應液回到室溫過濾不溶物,減壓濃縮濾液。以矽膠柱層析(己烷:乙酸乙酯=1:3)進行純化得到目的物。產量12mg(收率10%)。
1H-NMR(CDCl3,δ,ppm):5.56(2H,s),6.90(1H,td),7.32(1H,d),7.79(2H,m),7.84(1H,d),8.43(1H,d),8.56(1H,d)
MS:m/z=366(M+H)。
合成例34:N-[1-((6-氯吡啶-3-基)甲基)吡啶-2(1H)-亞基]-2,2-二氟乙醯胺(化合物8)的合成
將400mg(4.26mmol)2-氨基吡啶溶解於10ml無水二氯甲烷中,加入322μl(490mg、5.11mmol)二氟乙酸、982mg(5.10mmol)EDC-HCl、622mg(5.11mmol)DMAP,於室溫攪拌61小時。反應結束後,將反應液以二氯甲烷稀釋,以水洗滌1次、1%HCl水溶液洗滌2次後,以無水硫酸鎂乾燥,減壓濃縮得到2,2-二氟-N-(吡啶-2(1H)-亞基)乙醯胺102mg(收率14%)。
1H-NMR(CDCl3,δ,ppm):6.03(1H,t),7.15(1H,m),7.78(1H,td),8.20(1H,d),8.34(1H,dd),8.72(1H,brs)。
將100mg(0.58mmol)所述的方法所得到的2,2-二氟-N-(吡啶-2(1H)-亞基)乙醯胺溶解於10ml無水乙腈中,加入溶解於5ml無水乙腈的94mg(0.58mmol)2-氯-5-氯甲基吡啶,接著加入84mg(0.63mmol)碳酸鉀,加熱回流140分鐘。反應結束後,使反應液回到室溫,過濾除去不溶物,進行減壓濃縮。在其中加入醚而析出固體,因而進行濾取,充分乾燥得到目的物。產量63mg(收率37%)。
1H-NMR(CDCl3,δ,ppm):5.52(2H,s),5.90(1H,t),6.79(1H,td),7.33(1H,d),7.71(1H,m),7.77(1H,dd),7.85(1H,dd),8.45(1H,d),8.50(1H,d)
13C-NMR(DMSO-d6,δ,ppm):53.0,111.0(t),115.2,120.7,124.7,131.7,140.6,141.6,143.2,150.4,150.9,158.3,169.4(t)
MS:m/z=298(M+H)。
試驗例1
小菜蛾防除試驗
從槽栽培的捲心菜切下直徑5.0cm的葉盤,在其上散布調製成50%丙酮水(加用0.05%Tween20)的500ppm的式(I)所表示的化合物的藥液。風乾後,在其中放飼2齡幼蟲。然後,將其放置於25℃的恆溫室(16小時光照期-8小時黑暗期)。放飼3日後觀察蟲的生死,依據下式算出死蟲率。試驗通過2連制進行。
死蟲率(%)={死亡蟲數/(生存蟲數+死亡蟲數)}×100。
試驗例2
棉蚜防除試驗
從槽栽培的黃瓜切除直徑2.0cm的葉盤,在其上散布調製成50%丙酮水(加用0.05%Tween20)的500ppm的式(I)所表示的化合物的藥液。風乾後,在其中放飼1齡幼蟲。然後,將其放置於25℃的恆溫室(16小時光照期-8小時黑暗期)。放飼3日後觀察蟲的生死,依據下式算出死蟲率。試驗通過2連制進行。
死蟲率(%)={死亡蟲數/(生存蟲數+死亡蟲數)}×100。
試驗例3
灰飛蝨防除試驗
將播種48小時後的小麥苗根部,用200μL的調製成10%丙酮水的100ppm的式(I)所表示的化合物的藥液處理。72小時由根部吸收藥劑,然後,在其中放飼灰飛蝨的2齡幼蟲各10隻。然後,將其放置於25℃的恆溫室(16小時光照期-8小時黑暗期)。放飼4日後觀察蟲的生死,依據下式算出死蟲率。試驗通過2連制進行。
死蟲率(%)={死亡蟲數/(生存蟲數+死亡蟲數)}×100。
作為試驗例1~3的結果,表2顯示式(I)所表示的有害生物防除劑的具體的生物活性(死蟲率(%))。
表2
<對藥劑低靈敏性害蟲的效果>
參考例 褐飛蝨防除試驗
對槽栽培的稻苗,將調製成10%丙酮水的特定濃度的本發明化合物的藥液進行土壤灌溉處理。處理3日後,於稻苗上放飼靈敏性、或藥劑低靈敏性的褐飛蝨2齡幼蟲各10隻。然後,將其放置於25℃的恆溫室(16小時光照期-8小時黑暗期)。放飼3日後觀察蟲的生死,依據下式算出死蟲率。試驗通過2連制進行。
死蟲率(%)={死亡蟲數/(生存蟲數+死亡蟲數)}×100。
還有,試驗害蟲使用長是件室內累代飼育的蟲(敏感性系統)、(I)2007年在熊本縣內、(II)2005年在福岡縣內分別採集後,在室內累代飼育的蟲(野外採集系統)。
其結果顯示,化合物1對任一系統在0.05mg/苗的處理中死蟲率為100%,在0.005mg/苗的處理中死蟲率為90%以上。另外,化合物2在0.01mg/苗的處理中,死蟲率為(敏感性系統)72%,(II)70%,化合物19在0.01mg/苗的處理中,死蟲率為(敏感性系統)100%、(II)93%。另一方面,吡蟲啉在0.05mg/苗的處理中死蟲率為(敏感性系統)100%、(I)40%、(II)60%。
其結果顯示,化合物1對吡蟲啉低靈敏性的褐飛蝨,具有高的殺蟲活性。
參考例 灰飛蝨防除試驗
對槽栽培的稻苗,將調製成10%丙酮水的特定濃度的本發明化合物的藥液進行土壤灌溉處理。處理3日後,於稻苗上放飼靈敏性、或藥劑低靈敏性的褐飛蝨2齡幼蟲各10隻。然後,將其放置於25℃的恆溫室(16小時光照期-8小時黑暗期)。放飼3日後觀察蟲的生死,依據下式算出死蟲率。試驗通過2連制進行。
死蟲率(%)={死亡蟲數/(生存蟲數+死亡蟲數)}×100。
此外,試驗害蟲使用長期間在室內累代飼育的蟲(敏感性系統)、2006年在熊本縣內採集後,在室內累代飼育的蟲(野外採集系統)。
其結果顯示,化合物1對任一系統在0.01mg/苗的處理中死蟲率為100%,在0.005mg/苗的處理中死蟲率為90%以上。另外,化合物3在0.01mg/苗的處理中死蟲率為(敏感性系統)100%、(野外採集系統)90%。另一方面,吡蟲啉在0.01mg/苗的處理中死蟲率為(敏感性系統)100%、(野外採集系統)50%。另外,氟蟲腈在0.01mg/苗的處理中死蟲率為(敏感性系統)100%、(野外採集系統)70%。
該結果顯示,化合物1、3對吡蟲啉、及氟蟲腈低靈敏性的灰飛蝨具有高的殺蟲活性。
參考例 化合物1及吡蟲啉的使用家蠅粗酶提取液的體外代謝試驗
如Pest Management Science(2003),59(3),347-352、及Journal of Pesticide Science(2004),29(2),110-116所記載,已知吡蟲啉因受到氧化代謝而失活,認為是獲得抵抗性的機制之一。為了確認對獲得了這樣的抵抗性的害蟲的效果而進行以下的實驗。
對家蠅成蟲(0.645g),添加10mL的磷酸鉀緩衝液(pH7.4,含有1mM EDTA),用Physcotron均質機(日音醫理科器械製作所)充分磨碎。然後,以10,000g、15分鐘的條件,將磨碎物進行離心分離。將所得到的上清液進一步以100,000g、60分鐘的條件進行離心分離,得到沈澱物。將該沈澱物溶解於1mL的磷酸鉀緩衝液中,以此作為粗酶液使用。酶提取作業全部在冰上或4℃的條件下進行。
在1.5mL容量的管中以以下的比例混合試劑,在25℃使其反應40小時。反應後,加入1mL丙酮並攪拌,將所產生的沈澱以12000rpm離心5分鐘除去。餾去上清的丙酮,注入LC/MS進行分析。
上述粗酶提取液:300μL
化合物1的DMSO溶液:5μL
葡萄糖6磷酸溶液:5μL
NADP+溶液:5μL
葡萄糖6磷酸脫氫酶溶液:5μL
磷酸鉀緩衝液(pH7.4,含有1mM EDTA):180μL。
<分析條件>
柱:カプセルパックC18MG
移動相組成:
0~3分鐘:85%水、5%乙腈、10%甲酸水溶液(0.1v/v%)
3~30分鐘:85→25%水、5→65%乙腈、10%甲酸水溶液(0.1v/v%)
30.1~36分鐘:90%乙腈、10%甲酸水溶液(0.1v/v%)
柱溫:40℃流速:0.35mL/分鐘注入量:100μL
UV波長:化合物1:325nm;吡蟲啉:300nm。
其結果是,代謝物的面積%的合計,化合物1為0.08,吡蟲啉為2.55、與吡蟲啉相比,化合物1的代謝物的量非常少。由以上的結果顯示,即使對將吡蟲啉代謝失活的抵抗性害蟲,化合物1也可以有效地防除。
<對於動物寄生性害蟲的防除效果>
參考例 長角血蜱防除試驗
將本發明的化合物的200ppm、10ppm的丙酮溶液30μL加入4mL容量玻璃瓶中。將其載放於搖床上,在旋轉的同時進行風乾,而在瓶內壁形成化合物的幹膜。將瓶乾燥24小時以上,然後,在其中放飼10隻長角血蜱幼蟎,加蓋。將瓶靜置於25℃、溼度85%、全暗條件的恆溫室中。放飼1日後觀察其生死,依據下式算出死蟲率。試驗通過2連制進行。
死蟲率(%)={死亡蟲數/(生存蟲數+死亡蟲數)}×100。
其結果是,在200ppm的處理量,化合物1、化合物9顯示死蟲率80%以上的殺蟎效果。
在10ppm的處理量,化合物1、化合物9顯示死蟲率80%以上的高的殺蟎效果。
在同樣的試驗中,吡蟲啉在10ppm的處理量,死蟲率為4%。
參考例 小鼠體表的長角血蜱防除效果
將小鼠(ICR,雄性、5周齡)的背面體毛剃直徑約2cm,在該處將切至高度約1.5cm的15mL聚苯乙烯圓錐形管用瞬間粘接劑粘接。
將用以下的處方調製的有害生物防除劑的1000倍稀釋液20μL,滴加於粘接的試管內的小鼠體表。充分使其乾燥後,放飼10隻以上長角血蜱幼蟎在管內並加蓋。放飼3日後觀察長角血蜱的生死,依據下式算出吸血抑制率。
處方[液化滴劑]
化合物1 48重量%
乙醇 52重量%
將上述成分均勻地混合得到液化滴劑。
吸血抑制率(%)=100-{吸血蟎數/(生存蟎數+死亡蟎數)}×100。
其結果是,化合物1顯示吸血抑制率為91%這樣的長角血蜱防除效果。
產業可利用性
如以上所說明,根據本發明,可以根據需要一鍋有效且收率良好地製造作為有害生物防除劑有用的所述式(I)所表示的2-醯基亞氨基吡啶衍生物,進而,可以穩定且廉價地供給作為有害生物防除劑所要求的量。因此,本發明可以較大貢獻於有害生物防除的領域。