用於PSMA靶向的成像和放射療法的金屬/放射性金屬標記的PSMA抑制物的製作方法
2023-07-25 18:47:11 2
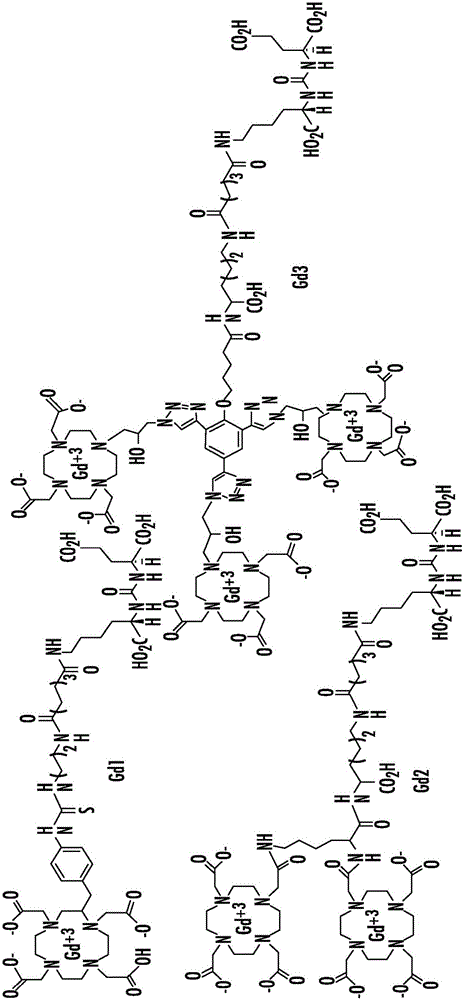
本申請要求2014年5月6日提交的美國臨時申請第61/989,428號和2015年2月18日提交的美國臨時申請第62/117,603號的權益,這些申請的每一個通過引用以其整體併入本文。聯邦政府資助的研究或開發本發明根據由美國國立衛生研究院(NationalInstituteofHealth)(NIH)授予的K25CA148901-01A1和U54CA1346751在政府支持下進行。政府在本發明中具有一定權利。背景前列腺特異性膜抗原(PSMA)日益地被認識到是用於前列腺和癌症的其他形式的成像和療法的可行的靶(Ghosh和Heston,2004;Milowsky等,2007;Olson等,2007)。PSMA在PCa和轉移灶,特別是對於激素難治性形式中顯著過表達(Ghosh和Heston,2004;Milowsky等,2007)。還已知PSMA由大部分實體瘤和腫瘤新血管系統表達(Haffner等,2012;Haffner等,2009)。成像PSMA可提供對雄激素信號傳導(Evans等,2011)和對紫杉烷類療法的響應(Hillier等,2011)的深刻理解。先前的研究已顯示了使用官能化的半胱氨酸-穀氨酸或賴氨酸-穀氨酸脲在前列腺癌的實驗模型中(Schulke等,2003;Mease等,2013;Banerjee等,2010)和在臨床上(Cho等,2012;Kulkarni等,2014;Zechmann等,2014)的PSMA靶向的放射性核素成像。對於大分子片段諸如放射性金屬(99mTc、68Ga、111In、86Y、203Pb、64Cu)複合物(Banerjee、Pullambhatla、Shallal,等,2011;Banerjee、Pullambhatla、Byun,等,2011;Banerjee等,2008)和納米粒子(Chandran等,2008;Kam等,2012)的附著,將長的接頭放置在大分子和靶向脲之間以保持PSMA-靶向的結合。不希望受任何一種特定的理論所束縛,由於配體結合位點的細胞外定位和估計的每細胞高受體濃度(~3.2μM/細胞體積),認為PSMA將是MR分子成像的適合的生物標誌物。MR成像為用於提供高解析度解剖和功能成像的臨床相關的、非侵入性診斷工具。分子MR成像使得生物標誌物能夠在體內可視化(Artemov、Mori、Okollie等,2003;Artemov、Mori、Ravi、Bhujwalla,等,2003;Konda等,2001;Lanza等,2004;Huang,等,2013)。由於它們易於施用並提供T1-加權的、正的對比,基於Gd(III)的造影劑被臨床醫師廣泛接受。儘管在具有高弛豫度的造影劑的設計中已取得了進展,但靈敏度仍然是分子MR成像的限制因素。對於在分子成像應用(特別地,用於成像受體或蛋白表達)中使用,基於Gd(III)-的造影劑很少超過檢測的限值(Artemov、Mori、Okollie等,2003;Artemov、Mori、Ravi、Bhujwalla,等,2003;Konda等,2001;Lanza等,2004;Huang,等,2013)。用信號放大策略,MR可能為與基於放射性核素的技術互補的分子成像提供靈敏模式(Aime等,2004;Major等,2009;Song等,2008;Artemov,2003)。儘管擴增策略可改進靶向的試劑的靈敏度,但從簡單的、低分子量化合物轉移至更大的、多重實體可顯著改變試劑的藥代動力學譜(Artemov、Mori、Okollie等,2003;Artemov、Mori、Ravi、Bhujwalla,等,2003;Konda等,2001;Lanza等,2004;Huang,等,2013)。Sherry等通過產生具有非常高的結合親和力(Kd)的造影劑解決了靈敏度的問題,使得通過MR檢測所需的試劑的量可最小化(Hanaoka等,2008;DeLeon-Rodriguez等,2010)。將受體特異性高親和力配體與用於檢測的多聚體Gd(III)試劑組合已經被設計為用於能夠基於MR的受體成像的一種解決方案(Wu等2012)。該方法的一個實例包括通過製備具有高縱向弛豫度(r1)值的多聚體Gd-樹狀體的VEGFR2的分子成像(DeLeon-Rodriguez等,2010)。已報導了在更高場強下具有改進的r1值的其他多聚體試劑,由於MR成像(實驗和臨床兩者)轉向更高的場(Mastarone2011)。在高場下優化弛豫度提供了較大的信噪比和對比噪聲比(SNR/CNR)的優點以及伴隨的較高空間解析度和降低獲取時間的優點(Rooney2007)。這些概念的組合,即高親和力靶向部分與靈敏多聚體造影劑的使用提供了研究表達前列腺特異性膜抗原(PSMA)的細胞和組織的靶向的MR成像的基本原理。此外,已推斷,基於脲的試劑也可用於含PSMA的病變的使用放射性核素的放射療法。事實上,使用該方法與[131I]MIP1095((S)-2-(3-((S)-1-羧基-5-(3-(4-[131I]碘苯基)脲基)戊基)脲基)戊二酸)(Zechmann等,2014)和177Lu-標記的PSMA-靶向的試劑(Kulkarni等2014)的臨床研究正在進行去勢抵抗性前列腺癌(castrate-resistantprostatecancer)的治療。這將類似於放射免疫療法(RIT),放射免疫療法(RIT)已證明在用常規地整合到臨床實踐中的兩種商業產品對淋巴瘤的治療中是非常成功的。然而,由於使用放射性標記的抗體用於成像,RIT充滿困難,包括延長的循環時間、不可預測的生物效應以及偶爾需要預靶向策略。此外,比起可在藥理學上操作的低分子量試劑,抗體可更少接近腫瘤。因此,對用於腫瘤的成像和放射療法的對PSMA具有高結合親和力的低分子量化合物仍存在需求。發射正電子的放射性核素86Y(半衰期[t1/2]=14.74h,β+=33%,Eβ+=664keV)是用於分子成像的有吸引力的同位素(Nayak和Brechbiel,2011)。釔-86可容易地採用86Sr(p,n)86Y核反應在小型生物醫學回旋加速器上製備(Yoo等,2005)。高能量β--發射體90Y(t1/2=64.06h,β-=72%,Eβ-=2.288MeV)用於腔內放射療法的廣泛使用(Witzig等,2003;Bodei等,2004)使86Y對於90Y-標記的放射療法的放射量測定法估計是理想的(Helisch等,2004)。用86Y放射性標記的抗體和肽具有與用90Y標記的那些相同的特性,使得對於用於放射療法的90Y的精確吸收劑量估計成為可能(Nayak和Brechbiel,2011;Palm等,2003)。由於177Lu和90Y具有相似的螯合化學反應,儘管177Lu具有比90Y短的β粒子範圍(t1/2=6.7天,Eβ-=0.5MeV),提出86Y作為適合的成像替代物以研究潛在的基於177Lu的放射療法以及用90Y放射性標記的那些。相似的基本原理已應用於神經內分泌靶向的肽受體放射性核素療法的試劑(Chen等,2012)。使用相似的方法,適合於SPECT成像的潛在的配對成像放射性同位素203Pb(半衰期,51.9h,Eβ-=279-keVγγ-射線,81%)可用於α-粒子療法的治療放射性核素212Pb(Chappell,等2000;Yong,等2011;Yong,等2012;Yong,等2013)。212Pb的衰變綱圖包括212Bi,212Bi在衰變後產生一個α-粒子、兩個ββ-粒子及數個γγ-發射。由於諸如局部緻密離子化的高線性能量轉移特性(其導致獨立於組織氧含量或劑量率的不可恢復的DNA雙鏈斷裂和細胞毒性),α-粒子發射體對於靶向的放射療法特別有吸引力(McDevitt,等,1998)。212Pb和212Bi都是有希望的α-粒子發射源,其具有充分描述的用於抗體連接的放射化學反應並由224Ra發生器容易地獲得。也已開發了在體外也顯示對PSMA的高結合親和力的基於放射性滷化氨基甲酸酯的PSMA抑制物,並且當用正電子發射體F-18進行放射性標記時,顯示在PSMA陽性小鼠腫瘤異種移植物中高攝取和從正常組織的快速清除。由於這類化合物的有利的藥代動力學譜,即,低非特異性結合、體內代謝不足和合理的腫瘤停留時間,成像研究已經擴展至分子放射療法。此外,基於氨基甲酸酯的抑制物可與金屬螯合劑採用與基於脲的金屬/基於放射性金屬的試劑相似的連接基官能性偶聯,以保持對PSMA的高結合親和力。因此,金屬或放射性金屬綴合的氨基甲酸酯支架也可用於表達PSMA的細胞和組織的成像和療法。概述在一些方面,本公開的主題提供了式(I)的化合物:其中:Z為四唑或CO2Q;Q為H或保護基團;X1和X2各自獨立地為NH或O;a為選自由1、2、3和4組成的組的整數;c為選自由0、1、2、3和4組成的組的整數;每個R1、R2和R4獨立地為H或C1-C4烷基;每個R3獨立地為H、C1-C6烷基或C2-C12芳基;W獨立地為O或S;Y為-NH-,並且可以存在或不存在;L為連接基,其中所述連接基選自由以下組成的組:其中:m為選自由1、2、3、4、5、6、7和8組成的組的整數;每個R5獨立地為H或–COOR6,其中每個R6獨立地為H或C1-C6烷基;n為選自由1、2、3、4、5、6、7、8、9、10、11和12組成的組的整數;p為選自由1、2、3、4、5、6、7和8組成的組的整數;Ch為可包含一種或更多種金屬或放射性金屬的螯合部分;或其藥學上可接受的鹽。在其他方面,本公開的主題提供了一種用於成像或治療一種或更多種前列腺特異性膜抗原(PSMA)腫瘤或細胞的方法,所述方法包括使一種或更多種腫瘤或細胞與有效量的式(I)的化合物接觸並製作圖像。上文已列舉了本公開的主題的某些方面,其全部或部分由本公開的主題所呈送,當結合下文作為最佳描述的所附實施例和附圖考慮時,其他方面隨著描述進行將變得明顯。附圖簡述如此以一般術語描述了本公開的主題,現將參考附圖,其不必然按比例繪製,且其中:圖1A和1B為:(A)Gd1、Gd2和Gd3的結構,和(B)IC50曲線;圖2示出了PC3PSMA-flu(藍色)和PC3PSMA+PIP(紅色)細胞沉澱中的Gd1-Gd3的濃度;數據從ICP-MS分析獲得;圖3示出了對於Gd1和Gd2,內化的和細胞表面結合的孵育劑量的百分比(%ID);數據從ICP-MS分析獲得;圖4A至圖4C示出了在同基因人類PC3前列腺癌細胞對,PSMA+PIP和PSMA-flu細胞中由Gd3產生的T1對比增強;(A)PIP和flu細胞的顏色編碼的T1圖。弛豫速率在9.4T在25℃下確定;(B)在用Gd3處理後,PIP和flu細胞中T1變化(ΔT1)的定量(n=4,P<0.05);(C)PIP和flu細胞中的Gd3的細胞攝取。與PIP細胞沉澱締合的Gd(III)的量顯著高於對於flu細胞沉澱的量。通過與ZJ43預孵育,PIP細胞中Gd3的累積被阻斷(n=4,P<0.05);圖5A至圖5D示出了(A)通過螢光成像的Gd1-Rh的細胞攝取和內化;PSMA+PC3PIP和PSMA-PC3flu細胞與Gd1-Rh的系列稀釋溶液(4μM–4nM)在37℃下孵育30min,隨後用冷PBS去除過量的造影劑;在4nM濃度的造影劑下的PC3PIP(B)和PC3flu(C)的放大圖;羅丹明螢光以紅色示出,且用DAPI染色的核計數器以藍色顯示;(D)Gd1-Rh的結構;圖6A和圖6B示出了在1h、4h和24h時的Gd3細胞表面結合(A)和內化(B)的%ID;圖7示出了與Gd1、Gd2、Gd3和Prohance孵育的PSMA-PC3flu細胞的存活力;造影劑與在多種Gd濃度下的細胞在37℃下孵育24小時,並且存活力使用MTS測定來測量;將存活力測量值對在任何造影劑不存在下生長的細胞歸一化;圖8示出了與Gd1、Gd2、Gd3和Prohance(Gd-DOTA)孵育的PSMA+PC3PIP細胞的存活力;造影劑與在多種Gd濃度下的細胞在37℃下孵育24小時,並且存活力使用MTS測定來測量;將存活力測量值對在任何造影劑不存在下生長的細胞歸一化;圖9A和圖9B示出了雄性NOD/SCID小鼠中的人類PC3前列腺癌PSMA+PIP和PSMA-flu腫瘤異種移植物的Gd3MR成像。(A)在將Gd3單次快速推注式注射(singlebolusinjection)到尾靜脈之後在40min、80min、120min和160min時在T2-加權成像後疊加PSMA+PC3PIP和PSMA-PC3flu腫瘤中的增強(ΔR1%)圖;(B)在將Gd3單次快速推注式注射到尾靜脈之後在40min、80min、120min和160min時無PSMA靶向部分的三聚體Gd造影劑的PSMA+腫瘤和PSMA-腫瘤中的ΔR1%圖;圖10A和圖10B示出了(A)在注射後1-1600min期間,對於每個腫瘤的整個體積計算的T1時間過程;和(B)在0-200min時的時間過程的放大區域;注意到在PSMA+PC3PIP腫瘤中的高特異性和持久性增強;圖11示出了在以0.05mmol/Kg劑量(n=3)注射Gd3之後對於小鼠的弛豫度的變化百分比(%ΔR1)。(p<0.03,PIP:flu);圖12示出了在注射1XPBS(磷酸鹽緩衝鹽水)之前和之後腫瘤(n=1)的T1值的體內時間依賴性變化;圖13A和圖13B示出了(A)圖11中示出的選擇的MR圖像;和(B)Gd3的結構;圖14示出了86Y-標記的PSMA抑制物的結構;圖15A和圖15B示出了[86Y]4的製備型HPLC色譜圖;(A)放射性-HPLC峰;和(B)在18.6min時的UV峰為在λ=254nm處的未螯合的4的;圖16A和圖16B示出了[86Y]5的製備型HPLC色譜圖;(A)放射性-HPLC峰,且(B)在34min時的UV峰為在λ=254nm處的未螯合的5的;圖17A至17C示出了[86Y]6的製備型HPLC色譜圖;(A)放射性-HPLC峰;(B)在15.8min時的UV峰為在λ=220nm處的未螯合的6的;和(C)純的[86Y]6的HPLC色譜圖;圖18A至18C示出了在注射後2h時攜帶PSMA+PC3PIP和PSMA-PC3flu腫瘤的小鼠中的(A)86Y-4、(B)86Y-5和(C)86Y-6的全身PET-CT成像。小鼠用~3.3mBq(90μCi)的放射性示蹤物靜脈內(IV)注射。PSMA+PC3PIP(實箭頭);PSMA-PC3flu(空箭頭);K=腎;GB=膽囊;GI=胃腸道;L=左側;R=右側。圖像被衰減校正和縮放至相同的最大值;圖19A和19B示出了攜帶PSMA+PC3PIP和PSMA-PC3flu腫瘤的小鼠中的[86Y]-4的PET-CT成像。(A)沒有和(B)使用有效的選擇性PSMA抑制物ZJ43作為阻斷劑(50mg/kg)阻斷PSMA獲得圖像。在用ZJ43共處理後,腫瘤和腎(另一PSMA+位點)兩者中放射性示蹤物攝取的降低提供了對PSMA特異性結合的進一步檢查。小鼠用~6.2MBq(168μCi)的放射性示蹤物IV注射。PSMA+PC3PIP(實箭頭);PSMA-PC3flu(空箭頭);K=腎;B=膀胱;L=左側;R=右側。圖像被衰減校正和縮放至相同的最大值;圖19C示出了有效的選擇性PSMA抑制物ZJ43的結構;圖20A至圖20C示出了在(A)注射後0.5h、(B)注射後2h和(C)注射後12h時攜帶PSMA+PC3PIP和PSMA-PC3flu腫瘤的小鼠中的86Y-6的PET-CT成像。小鼠用~6.2MBq(160μCi)的放射性示蹤物IV注射。PSMA+PC3PIP(實箭頭);PSMA-PC3flu(空箭頭);K=腎;L=左側;R=右側。圖像被衰減校正和縮放至相同的最大值;圖21A和圖21B示出了在(A)注射後1-2h和(B)注射後2-3.5h時狒狒中的86Y-6PET的3D時間過程MIP(最大強度再投影)展示。為了增強可視化,膀胱放射性使用閾值方法半自動地分割並隨後被去除。採用MIP3D渲染來提供全身放射性示蹤物分布的概述。在除了膀胱(未示出)和腎(K)以外的大部分正常組織中觀察到很少的放射性示蹤物。將動物插入導管用於該研究。注意到淚腺、腮腺和唾液腺中的輕度攝取(分別為短、長和空的箭頭);圖22示出了177Lu-SRV171和進一步改進體內藥代動力學的相關的提議的試劑的結構;圖23示出了在37℃下2h之後177Lu-SRV171(0.01-10μCi/百萬個PSMA+PIP和PSMA-flu細胞)的孵育劑量(ID)的百分比。攝取特異性通過10μMZJ43的共孵育進一步檢查;圖24示出了多達24h的177Lu-SRV171(1μCi)的內化研究;圖25A至圖25C示出了在(A)注射後2h、(B)注射後24h和(C)注射後96h時,使用177Lu-SRV171(500μCi)的攜帶PIP和flu腫瘤的雄性小鼠的SPECT圖像。在腎(K)、膀胱(B)和flu腫瘤中發現低攝取。圖26示出了在注射後3h、24h、48h、72h和96h時的不同器官中的177Lu-SRV171的組織生物分布;圖27示出了203Pb-SR-IX-11和203Pb-SRV171的結構;圖28A至圖28C示出了在(A)注射後60min、(B)注射後120min和(C)注射後240min時,使用203Pb-SRV171(左側)和203Pb-SR-IX-11(右側)的攜帶PIP和flu腫瘤的雄性小鼠的SPECT-CT圖像;圖29示出了用於設計本公開的主題的化合物的兩種賴氨酸-氨基甲酸酯支架:對應於氨基甲酸酯支架的氧戊二酸(oxypentanedioicacid)(OPA)和對應於「反」氨基甲酸酯支架的氨基戊二酸(NPA);圖30示出了ZCP-01的HPLC色譜圖;圖31示出了冷的[In]ZPC-01的電噴霧電離質譜(ESI-MS);圖32示出了冷的[In]ZPC-01的HPLC色譜圖;圖33A和圖33B示出了[In]ZCP-01的製備型HPLC色譜圖;(A)放射性-HPLC峰;且(B)在32min時的UV峰為在λ=200nm處的未螯合的ZCP-01的;以及圖34A至34C示出了在注射之後(A)2h、(B)4h和(C)24h,攜帶PSMA+PC3Pip和PSMA-PCflu腫瘤異種移植物的小鼠中的[111In]ZCP-01的攝取。詳細描述本公開的主題現將參考所附實施例和附圖在下文更充分地被描述,其中本公開的主題的一些而非所有實施方案被示出。本公開的主題可以以許多不同的形式來體現並且不應該被解釋為限於本文列出的實施方案;相反,提供這些實施方案使得本公開內容將滿足適用的法律要求。事實上,本文闡述的本公開的主題的許多修改形式和其他實施方案將為公開的主題所屬
技術領域:
的技術人員想到具有上述說明和相關實施例和附圖中呈現的教導的益處。因此,應該理解,本公開的主題不限於公開的具體實施方案並且修改形式和其他實施方案預期被包括在所附權利要求書的範圍內。I.用於PSMA靶向的成像和放射療法的金屬/放射性金屬標記的PSMA抑制物磁共振(MR)成像由於可同時提供解剖、功能和分子信息是有利的。MR分子成像可將這種已確立的臨床模式及其高空間解析度的普遍存在與體內分子譜系分析相結合。然而,由於MR的固有的低靈敏度,需要高的局部濃度的生物靶以產生可辨別的MR對比。不希望受任何一種特定理論所束縛,認為,由於每細胞的高靶濃度(約3μM/細胞體積)以及配體結合位點的細胞外定位,PSMA將是用於MR分子顯像劑的良好靶。本公開的方法針對改進造影劑對特定分子或細胞靶的結合親和力(最低Kd),使得MR檢測所需的試劑的量將少得多。因此,本公開的方法將高結合親和力受體特異性配體與多聚體Gd(III)試劑組合作為用於基於MR的分子成像的一種可能的解決方案。先前,在小鼠中使用放射性標記的、基於脲的PSMA抑制物顯示成功的基於放射性金屬的PET(64Cu)和SPECT(111In和99mTc)成像。開發了包含以下的三重策略:(i)PSMA靶向部分,(ii)用於藥代動力學調節的連接基,及(iii)使得能夠附連放射性核素的螯合劑。該策略包括用於PET成像的86Y標記的DOTA綴合的試劑,並且為了用作放射療法的模型,帶有對應的90Y標記的試劑。由於DOTA為用於許多金屬的強螯合劑,相同的DOTA綴合物可與其他放射治療放射性核素一起使用,其他放射治療放射性核素諸如Lu-177、Ac-225、Bi-213、Bi-212、Pb-212、Cu-67和Sc-47。在本公開的主題中,使用相同的脲-連接基構建體,並且增加Gd-螯合物(單體-Gd、二聚體-Gd和三聚體Gd)的數目以優化如高場造影劑的弛豫行為或MR靈敏度以及它們的結合親和力,以系統地研究PCa的基於PSMA的MR成像的可能性。A.式(I)的化合物因此,在一些實施方案中,本公開的主題提供了式(I)的化合物:其中:Z為四唑或CO2Q;Q為H或保護基團;X1和X2各自獨立地為NH或O;a為選自由1、2、3和4組成的組的整數;c為選自由0、1、2、3和4組成的組的整數;每個R1、R2和R4獨立地為H或C1-C4烷基;每個R3獨立地為H、C1-C6烷基或C2-C12芳基;W獨立地為O或S;Y為-NH-,並且可以存在或不存在;L為連接基,選自由以下組成的組:其中:m為選自由1、2、3、4、5、6、7和8組成的組的整數;每個R5獨立地為H或–COOR6,其中每個R6獨立地為H或C1-C6烷基;n為選自由1、2、3、4、5、6、7、8、9、10、11和12組成的組的整數;p為選自由1、2、3、4、5、6、7和8組成的組的整數;Ch為可包含一種或更多種金屬或放射性金屬的螯合部分;或其藥學上可接受的鹽。式(I)不包括在WO2009/002529、WO2010/108125和WO2013/082338中公開的化合物,特別地,以下化合物明確地從本申請的產品權利要求(matterclaims)的組合物中排除:在更特定的實施方案中,螯合部分選自由以下組成的組:其中q為選自由1、2、3、4、5、6、7和8組成的組的整數。在又更特定的實施方案中,式(I)的化合物選自由以下組成的組:其中:x選自由2和3組成的組:M為金屬或放射性金屬;或其藥學上可接受的鹽。在一些實施方案中,金屬選自由以下組成的組:Gd、Lu、Ac、Bi、Pb、Cu、In、Sc和Y。在特定的實施方案中,金屬或放射性金屬選自由以下組成的組:Gd-157、Lu-177、Ac-225、Bi-212、Bi-213、Pb-203/Pb-212、Cu-67、In-111、Sc-44/Sc-47和Y-90。在又更特定的實施方案中,對於MRI應用,非放射性金屬為Gd-157(穩定同位素);對於放射療法應用,放射性金屬選自由Lu-177、Ac-225、Bi-203、Pb-210、Cu-67、In-111、Sc-47和Y-90組成的組;對於PET成像,放射性金屬選自由Y-86和Sc-44組成的組;且對於SPECT應用,放射性金屬選自由Lu-177和In-111組成的組。B.使用式(I)的化合物用於MR成像和/或治療表達PSMA的腫瘤或細胞的方法在一些實施方案中,本公開的主題提供了一種用於成像或治療一種或更多種前列腺特異性膜抗原(PSMA)腫瘤或細胞的方法,所述方法包括使一種或更多種腫瘤或細胞與有效量的式(I)的化合物接觸並製作圖像,所述式(I)的化合物包括:其中:Z為四唑或CO2Q;Q為H或保護基團;X1和X2各自獨立地為NH或O;a為選自由1、2、3和4組成的組的整數;c為選自由0、1、2、3和4組成的組的整數;每個R1、R2、R3和R4獨立地為H或C1-C4烷基;W獨立地為O或S;Y為-NH-,並且可以存在或不存在;L為連接基,選自由以下組成的組:其中:m為選自由1、2、3、4、5、6、7和8組成的組的整數;每個R5獨立地為H或–COOR6,其中每個R6獨立地為H或C1-C6烷基;n為選自由1、2、3、4、5、6、7、8、9、10、11和12組成的組的整數;p為選自由1、2、3、4、5、6、7和8組成的組的整數;Ch為螯合部分,可包含一種或更多種金屬或放射性金屬;或其藥學上可接受的鹽。「接觸」意指導致本公開的主題的包含顯像劑的至少一種化合物物理接觸至少一種表達PSMA的腫瘤或細胞的任何行動。接觸可包括使一種或更多種細胞或一種或更多種腫瘤暴露於足以導致至少一種化合物與至少一種細胞或腫瘤接觸的量的化合物。該方法可通過如下體外或離體被實踐:通過在受控環境諸如培養皿或管中引入,且優選地混合化合物和一種或更多種細胞或一種或更多種腫瘤。該方法可在體內實踐,在該情況下接觸意味著使受試者中的至少一種細胞或腫瘤暴露於本公開的主題的至少一種化合物,諸如經由任何適合的途徑將化合物施用至受試者。根據本公開的主題,接觸可包括在遠離待接觸的細胞的位點使化合物引入、暴露等,並允許受試者的身體機能,或天然(例如,擴散)或人為引入(例如,旋轉)的流體運動,導致化合物和一種或更多種細胞或一種或更多種腫瘤接觸。在一些實施方案中,腫瘤或細胞見於體外、體內或離體。「製作圖像」意指使用基於磁共振(MR)(極化和激發組織中的水分子中的氫核以產生可檢測的信號的磁體),以形成細胞、組織、腫瘤、身體的部分等的圖像。式(I)不包括在WO2009/002529、WO2010/108125和WO2013/082338中公開的化合物,特別地,以下化合物明確地從本申請的成像權利要求中排除:在更特定的實施方案中,螯合部分選自由以下組成的組:其中q為選自由1、2、3、4、5、6、7和8組成的組的整數。在又更特定的實施方案中,化合物選自由以下組成的組:x選自由2和3組成的組:M為金屬或放射性金屬;或其藥學上可接受的鹽。在一些實施方案中,金屬選自由以下組成的組:Gd、Lu、Ac、Bi、Pb、Cu、In、Sc和Y。在特定的實施方案中,金屬或放射性金屬選自由以下組成的組:Gd-157、Lu-177、Ac-225、Bi-203、Pb-210、Cu-67、In-111、44Sc-/47Sc和Y-90。在又更特定的實施方案中,對於MRI應用,非放射性金屬為Gd-157(穩定同位素);對於放射療法應用,放射性金屬選自由Lu-177、Ac-225、Bi-203、Pb-210、Cu-67、In-111、Sc-47和Y-90組成的組;對於PET成像,放射性金屬選自由Y-86和Sc-44組成的組;且對於SPECT應用,放射性金屬選自由Lu-177和In-111組成的組。在某些實施方案中,一種或更多種表達PSMA的腫瘤或細胞選自由以下組成的組:前列腺腫瘤或細胞、轉移的前列腺腫瘤或細胞、肺腫瘤或細胞、腎腫瘤或細胞、成膠質細胞瘤、胰腺腫瘤或細胞、膀胱腫瘤或細胞、肉瘤、黑素瘤、乳腺腫瘤或細胞、結腸腫瘤或細胞、生殖細胞、嗜鉻細胞瘤、食管腫瘤或細胞、胃腫瘤或細胞、及其組合。在又更多的某些實施方案中,一種或更多種表達PSMA的腫瘤或細胞為前列腺腫瘤或細胞。在一些實施方案中,一種或更多種表達PSMA的腫瘤或細胞是體外、體內或離體的。在特定的實施方案中,一種或更多種表達PSMA的腫瘤或細胞存在於受試者中。在一些實施方案中,腫瘤或細胞見於受試者中。通過本公開的方法在其許多實施方案中治療的受試者期望地為人受試者,儘管應該理解,本文描述的方法關於所有脊椎動物物種是有效的,所述所有脊椎動物物種旨在被包括在術語「受試者」中。因此,「受試者」可包括用於醫學目的,諸如用於現存狀況或疾病的治療或用於防止狀況或疾病的發作的預防性治療的人類受試者,或用於醫學目的、獸醫目的或發育目的的動物(非人類)受試者。適合的動物受試者包括哺乳動物,哺乳動物包括但不限於,靈長類動物(primates),例如,人、猴、猿等;牛科動物(bovine),例如,家牛(cattle)、公牛(oxen)等;綿羊類(ovine),例如,綿羊(sheep)等;山羊類(caprine),例如,山羊(goat)等;豬類(porcines),例如,豬、肉豬(hogs)等;馬科動物(equines),例如,馬、驢、斑馬等;貓科動物(feline),包括野生貓和家養貓;犬類(canine),包括狗;兔類動物(lagomorph),包括家兔、野兔等;和嚙齒類動物(rodent),包括小鼠、大鼠等。動物可以為轉基因動物。在一些實施方案中,受試者為人類,包括但不限於,胎兒、新生兒、嬰兒、青少年和成年受試者。此外,「受試者」可包括患有或懷疑患有狀況或疾病的患者。因此,術語「受試者」和「患者」在本文可互換地使用。在一些實施方案中,受試者為人類。在其他實施方案中,受試者為非人類的。在一些實施方案中,將可檢測有效量的本公開的方法的顯像劑施用至受試者。根據本公開的主題,顯像劑的「可檢測有效量」被定義為使用可用於臨床使用的設備足以產生可接受的圖像的量。可檢測有效量的顯像劑可以多於一次注射施用。顯像劑的可檢測有效量可根據以下因素而變化:諸如個體的易感性程度,個體的年齡、性別和體重,個體的特異性響應,劑量學,以及儀器和膠片相關因素。此類因素的優化完全在本領域的技術水平內。優選的是,本公開的主題的化合物從身體的組織快速排洩。通常,本公開的主題的化合物在小於約24小時內從身體排出。更優選地,本公開的主題的化合物在小於約16小時、12小時、8小時、6小時、4小時、2小時、90分鐘或60分鐘內從身體排出。在一些實施方案中,本公開的方法包括從受試者的腫瘤或細胞清除包含顯像劑的化合物。本公開的方法的至少一個優點是,在一些實施方案中,包含顯像劑的化合物從受試者的腎比從腫瘤更快速清除。在一些實施方案中,本公開的方法使用在體內穩定的化合物,使得基本上所有,例如,多於約50%、60%、70%、80%或更優選地90%的注射的化合物在排洩之前不被身體代謝。在其他實施方案中,包含顯像劑的化合物在體內是穩定的。C.定義i.化學定義儘管關於式(I)的化合物的以下術語被認為是本領域普通技術人員充分理解的,闡述以下定義以便於解釋本公開的主題。這些定義旨在補充和說明而非排除對於本領域普通技術人員在閱讀本公開內容之後將明顯的定義。如本文使用的,無論是否前面加上術語「任選地」,術語被取代的(substituted)和取代基(substituent)指如該領域技術人員所理解的,將一個官能基團改變為另一官能基團的能力,條件是保持所有原子的化合價。當任何給定結構中的多於一個位置可被選自指定組的多於一個取代基取代時,取代基在每個位置處可相同或不同。取代基還可被進一步取代(例如,芳基基團取代基可被另一個取代基,諸如另一個芳基基團取代,該另一個芳基基團在一個或更多個位置處被,例如,氟進一步取代)。當取代基基團或連接基團由其從左至右書寫的常規化學式指定時,它們同樣包括由從右至左書寫結構而產生的化學上相同的取代基,例如,-CH2O-等同於-OCH2-;-C(=O)O-等同於-OC(=O)-;-OC(=O)NR-等同於-NRC(=O)O-,等。如本文使用的,當內部取代基側翼為鍵(例如,-NRC(O)-)時,原子的順序是固定的,基團的定向可不翻轉,且以呈現的定向插入到結構中。換言之,-NRC(O)-與-C(O)NR-是不同的。如本文使用的術語C(O)(例如-NRC(O)-)用於指示羰基(C=O)基團,其中氧通過雙鍵與碳鍵合。當使用術語「獨立地選擇的」時,被提及的取代基(例如,R基團,諸如基團R1、R2等,或變量,諸如「m」和「n」)可相同或不同。例如,R1和R2兩者可以是被取代的烷基,或者R1可以是氫,且R2可以是被取代的烷基,等。當提及本文中的取代基的組使用時,術語「一(a)」、「一(an)」或「一(一)(a(n))」是指至少一個。例如,當化合物被「一個」烷基或芳基取代時,該化合物任選地被至少一個烷基和/或至少一個芳基取代。此外,當部分被R取代基取代時,該基團可被稱為「R-取代的」。當部分為R-取代的時,該部分被至少一個R取代基取代,且每個R取代基任選地不同。命名的「R」或基團將通常具有在本領域中公認為對應於具有該名稱的基團的結構,除非本文另有規定。出於說明的目的,下文定義如以上列出的某些代表性的「R」基團。本公開內容的化合物的描述受本領域技術人員已知的化學鍵合原理的限制。因此,當基團可被許多取代基中的一個或更多個取代時,選擇此類取代以符合化學鍵合的原理,並得到並非固有不穩定的和/或對於本領域普通技術人員將已知在環境條件,諸如水性、中性和幾種已知的生理條件下可能不穩定的化合物。例如,遵照本領域技術人員已知的化學鍵合原理,雜環烷基或雜芳基經由環雜原子附連至分子的剩餘部分,從而避免固有不穩定的化合物。如本文使用的術語烴指包含氫和碳的任何化學基團。烴可以是被取代的或未被取代的。如本領域技術人員將已知的,在進行任何取代時必須滿足所有化合價。烴可以是不飽和的、飽和的、支鏈的、無支鏈的、環狀的、多環的或雜環的。說明性烴在本文下文進一步定義並且包括,例如,甲基、乙基、正丙基、異丙基、環丙基、烯丙基、乙烯基、正丁基、叔丁基、乙炔基、環己基、甲氧基、二乙基氨基等。除非另有說明,術語「烷基」本身或作為另一取代基的一部分意指直鏈(即,無支鏈)或支鏈、無環或環狀烴基團或其組合,其可以是完全飽和的、單不飽和的或多不飽和的,並且可包括具有指定碳原子數目(即,C1-C10意指1個至10個碳)的二價基團和多價基團。在特定的實施方案中,術語「烷基」指包含C1-20的線性(linear)(即,「直鏈(straight-chain)」)、支鏈或環狀的、飽和或至少部分不飽和的且在一些情況下完全不飽和的(即,烯基和炔基)烴基,其通過去除單個氫原子來源於包含介於1個和20個之間碳原子的烴部分。代表性的飽和烴基團包括,但不限於,甲基、乙基、正丙基、異丙基、正丁基、異丁基、仲丁基、叔丁基、正戊基、仲戊基、異戊基、新戊基、正己基、仲己基、正庚基、正辛基、正癸基、正十一烷基、十二烷基、環己基、(環己基)甲基、環丙基甲基及其同系物和異構體。「支鏈的」指其中低級烷基基團,諸如甲基、乙基或丙基被附連至線性烷基鏈上的烷基基團。「低級烷基」指具有1個至約8個碳原子(即,C1-8烷基),例如,1個、2個、3個、4個、5個、6個、7個或8個碳原子的烷基。「高級烷基」指具有約10個至約20個碳原子,例如,10個、11個、12個、13個、14個、15個、16個、17個、18個、19個或20個碳原子的烷基。在某些實施方案中,「烷基」特別指C1-8直鏈烷基。在其他實施方案中,「烷基」特別指C1-8支鏈烷基。在某些實施方案中,烷基基團為C1-C6烷基基團或C1-C4烷基基團。如本文使用的術語「C1-C6烷基」意指完全飽和的直鏈、支鏈或環狀C1-C6烴及其雜化物,諸如(環烷基)烷基。C1-C6烷基取代基的實例包括甲基(Me)、乙基(Et)、丙基(包括正丙基(n-Pr,nPr)、異丙基(i-Pr,1Pr)、和環丙基(c-Pr,0Pr))、丁基(包括正丁基(n-Bu,nBu)、異丁基(i-Bu,1Bu)、仲丁基(s-Bu,sBu)、叔丁基(t-Bu,1Bu)、或環丁基(c-Bu,0Bu)),等等。烷基基團可任選地被一個或更多個烷基基團取代基取代(「取代的烷基」),所述取代基可相同或不同。術語「烷基基團取代基」包括但不限於烷基、被取代的烷基、滷代、芳基氨基、醯基、羥基、芳氧基、烷氧基、烷基硫代、芳基硫代、芳基烷基氧基、芳基烷基硫代、羧基、烷氧基羰基、氧代和環烷基。可沿著烷基鏈任選地插入一個或更多個氧、硫、或被取代或未被取代的氮原子,其中氮取代基為氫、低級烷基(本文中也稱為「烷基氨基烷基」)、或芳基。因此,如本文使用的,術語「被取代的烷基」包括如本文定義的烷基基團,其中烷基基團的一個或更多個原子或官能基團被另一個原子或官能基團代替,所述另一個原子或官能基團包括例如,烷基、被取代的烷基、滷代、芳基、被取代的芳基、烷氧基、羥基、硝基、氨基、烷基氨基、二烷基氨基、硫酸酯基(sulfate)和巰基。除非另有說明,術語「雜烷基」本身或與另一術語組合意指由至少一個碳原子和選自由O、N、P、Si及S組成的組的至少一個雜原子組成的穩定的直鏈或支鏈或環狀烴基團或其組合,並且其中氮、磷和硫原子可任選地被氧化,且氮雜原子可以任選地被季銨化。一個或更多個雜原子O、N、P以及S和Si可位於雜烷基基團的任何內部位置處或在烷基基團被附連至分子的剩餘部分的位置處。實例包括,但不限於,-CH2-CH2-O-CH3、-CH2-CH2-NH-CH3、-CH2-CH2-N(CH3)-CH3、-CH2-S-CH2-CH3、-CH2-CH25-S(O)-CH3、-CH2-CH2-S(O)2-CH3、-CH=CH-O-CH3、-Si(CH3)3、-CH2-CH=N-OCH3、-CH=CH-N(CH3)-CH3、0-CH3、-0-CH2-CH3、及-CN。多達兩個或三個雜原子可以是連續的,諸如,例如,-CH2-NH-OCH3和-CH2-O-Si(CH3)3。如以上描述的,如本文使用的雜烷基基團包括通過雜原子附連至分子剩餘部分的那些基團,諸如-C(O)R』、-C(O)NR』、-NR』R」、-OR』、-SR、和/或-SO2R』。當敘述「雜烷基」,隨後敘述具體的雜烷基基團(諸如-NR』R等)時,應當理解,術語雜烷基和-NR』R」不是冗餘的或相互排斥的。而是,敘述具體的雜烷基基團以增加清楚性。因此,術語「雜烷基」在本文中不應該被解釋為排除具體的雜烷基基團,諸如-NR'R」等。在術語「(環烷基)烷基」中,環烷基和烷基如以上定義的,且附連點在烷基基團上。該術語包括,但不限於,環丙基甲基、環戊基甲基和環己基甲基。烷基可以是被取代的或未被取代的。「環狀的」和「環烷基」指約3個至約10個碳原子,例如,3個、4個、5個、6個、7個、8個、9個、或10個碳原子的非芳族單環或多環系統。環烷基基團可任選地是部分不飽和的。環烷基基團還可任選地被如本文定義的烷基基團、氧代和/或亞烷基取代。可沿著環狀烷基鏈任選地插入一個或更多個氧、硫、或被取代或未被取代的氮原子,其中氮取代基為氫、烷基、被取代的烷基、芳基或被取代芳基,由此提供雜環基團。代表性的單環狀環烷基環包括環丙基、環丁基、環戊基、環己基和環庚基。多環狀環烷基環包括金剛烷基、八氫萘基(octahydronaphthyl)、十氫萘(decalin)、樟腦(camphor)、莰烷(camphane)和正金剛烷基(noradamantyl)、以及稠環體系,諸如二氫萘和四氫萘(tetrahydronaphthalene)等。術語「環雜烷基」或「雜環烷基」指包括一個或更多個雜原子的非芳族環系統、不飽和或部分不飽和的環系統,諸如3元至10元被取代或未被取代的環烷基環系統,並且任選地可包括一個或更多個雙鍵,所述雜原子可相同或不同,且選自由氮(N)、氧(O)、硫(S)、磷(P)和矽(Si)組成的組。環雜烷基環可任選地被稠合至或以其他方式附連至其他環雜烷基環和/或非芳族烴環。雜環狀環包括具有1個至3個獨立地選自氧、硫和氮的雜原子的那些環,其中氮和硫雜原子可任選地被氧化,並且氮雜原子可任選地被季銨化。在某些實施方案中,術語雜環指非芳族5-元、6-元或7-元環或多環狀基團,其中至少一個環原子為選自O、S和N的雜原子(其中氮和硫雜原子可任選地被氧化),包括,但不限於,雙環狀或三環狀基團,包括具有介於一個和三個之間獨立地選自氧、硫和氮的雜原子的稠合六元環,其中(i)每個5-元環具有0至2個雙鍵,每個6-元環具有0至2個雙鍵,且每個7-元環具有0至3個雙鍵,(ii)氮和硫雜原子可任選地被氧化,(iii)氮雜原子可任選地被季銨化,以及(iv)任何以上雜環狀環可與芳基或雜芳基環稠合。代表性的環雜烷基環系統包括,但不限於吡咯烷基、吡咯啉基、咪唑烷基、咪唑啉基、吡唑烷基、吡唑啉基、哌啶基、哌嗪基、二氫吲哚基、奎寧環基、嗎啉基、硫代嗎啉基(thiomorpholinyl)、噻二嗪基(thiadiazinanyl)、四氫呋喃基(tetrahydrofuranyl)等。除非另有說明,術語「環烷基」和「雜環烷基」本身或與其他術語組合分別表示「烷基」和「雜烷基」的環狀形式。另外,對於雜環烷基,雜原子可佔據雜環被附連至分子的剩餘部分處的位置。環烷基的實例包括,但不限於,環戊基、環己基、1-環己烯基、3-環己烯基、環庚基等。雜環烷基的實例包括,但不限於,1-(1,2,5,6-四氫吡啶基)、1-哌啶基、2-哌啶基、3-哌啶基、4-嗎啉基、3-嗎啉基、四氫呋喃-2-基、四氫呋喃-3-基、四氫噻吩-2-基、四氫噻吩-3-基、1-哌嗪基、2-哌嗪基等。術語「亞環烷基」和「雜亞環烷基」分別指環烷基和雜環烷基的二價衍生物。如本文使用的術語「環烷基烷基」指如以上定義的環烷基基團,其通過也如以上定義的烷基基團附連至母體分子部分。環烷基烷基的實例包括環丙基甲基和環戊基乙基。不飽和烷基基團為具有一個或更多個雙鍵或三鍵的烷基基團。不飽和烷基基團的實例包括,但不限於,乙烯基、2-丙烯基、丁烯基、2-異戊烯基、2-(丁二烯基)、2,4-戊二烯基、3-(1,4-戊二烯基)、乙炔基、1-丙炔基和3-丙炔基、3-丁炔基(3-butynyl)、以及更高級的同系物和異構體。限於烴基團的烷基基團被稱為「同烷基(homoalkyl)」。更具體地,如本文使用的術語「烯基」指通過去除單個氫原子具有至少一個碳-碳雙鍵的來源於包含C1-20的直鏈或支鏈烴部分的單價基團。烯基基團包括,例如,乙烯基(ethenyl)(即,乙烯基(vinyl))、丙烯基、丁烯基、1-甲基-2-丁烯-1-基、戊烯基、己烯基、辛烯基和丁二烯基。如本文使用的術語「環烯基」指包含至少一個碳-碳雙鍵的環狀烴。環烯基基團的實例包括環丙烯基、環丁烯基、環戊烯基、環戊二烯、環己烯基、1,3-環己二烯、環庚烯基、環庚三烯基和環辛烯基。如本文使用的術語「炔基」指包含至少一個碳-碳三鍵的指定碳原子數目的來源於直鏈或支鏈C1-20烴的一價基團。「炔基」的實例包括乙炔基、2-丙炔基(2-propynyl)(炔丙基(propargyl))、1-丙炔基、戊炔基(pentynyl)、己炔基(hexynyl)、庚炔基(heptynyl)和丙二烯基基團(allenylgroup)等。術語「亞烷基」本身或作為另一取代基的部分指來源於具有從1個至約20個碳原子的烷基基團的直鏈或支鏈二價脂族烴基團,從1個至約20個碳原子例如1個、2個、3個、4個、5個、6個、7個、8個、9個、10個、11個、12個、13個、14個、15個、16個、17個、18個、19或20個碳原子。亞烷基基團可以是直鏈、支鏈或環狀的。亞烷基基團還可任選地是不飽和的和/或被一個或更多個「烷基基團取代基」取代。可沿著亞烷基基團任選地插入一個或更多個氧、硫或被取代或未被取代的氮原子(本文中也稱為「烷基氨基烷基」),其中氮取代基為如先前描述的烷基。示例性亞烷基基團包括亞甲基(–CH2–);乙烯(–CH2–CH2–);丙烯(–(CH2)3–);環亞己基(–C6H10–);–CH=CH–CH=CH–;–CH=CH–CH2–;-CH2CH2CH2CH2-,-CH2CH=CHCH2-,-CH2CsCCH2-,-CH2CH2CH(CH2CH2CH3)CH2-,–(CH2)q–N(R)–(CH2)r–,其中q和r的每個獨立地為從0至約20的整數,例如,0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20,且R為氫或低級烷基;亞甲基二氧基(methylenedioxyl)(–O–CH2–O–);和亞乙基二氧基(ethylenedioxyl)(–O–(CH2)2–O–)。亞烷基可具有約2個至約3個碳原子且還可具有6-20個碳。通常,烷基(或亞烷基)基團將具有從1個至24個碳原子,其中具有10個或更少碳原子的那些基團是本公開內的一些實施方案。「低級烷基」或「低級亞烷基」為較短鏈的烷基或亞烷基基團,通常具有8個或更少的碳原子。術語「雜亞烷基」本身或作為另一取代基的一部分意指來源於雜烷基的二價基團,作為例示,但不限於,-CH2-CH2-S-CH2-CH2-和-CH2-S-CH2-CH2-NH-CH2-。對於雜亞烷基,雜原子也可佔據鏈末端的任一個或兩個(例如,亞烷基氧代、亞烷基二氧代、亞烷基氨基、亞烷基二氨基等)。更進一步,對於亞烷基和雜亞烷基連接基團,連接基團的定向不暗示在其連接基團的式被書寫的方向。例如,式-C(O)OR』-表示-C(O)OR』-和–R』OC(O)-兩者。除非另有說明,術語「芳基」意指芳族烴取代基,其可以是單環或稠合在一起或共價連接的多環(諸如從1個至3個環)。術語「雜芳基」指包含從1個至4個選自N、O和S的雜原子(在多個環的情況下在每個單獨的環中)的芳基基團(或環),其中氮和硫原子任選地被氧化,並且一個或更多個氮原子任選地被季銨化。雜芳基基團可通過碳或雜原子附連至分子的剩餘部分。芳基和雜芳基的非限制性實例包括苯基、1-萘基、2-萘基、4-聯苯基、1-吡咯基、2-吡咯基、3-吡咯基、3-吡唑基、2-咪唑基、4-咪唑基、吡嗪基、2-噁唑基、4-噁唑基、2-苯基-4-噁唑基、5-噁唑基、3-異噁唑基、4-異噁唑基、5-異噁唑基、2-噻唑基、4-噻唑基、5-噻唑基、2-呋喃基、3-呋喃基、2-噻吩基、3-噻吩基、2-吡啶基、3-吡啶基、4-吡啶基、2-嘧啶基、4-嘧啶基、5-苯並噻唑基、嘌呤基、2-苯並咪唑基、5-吲哚基、吲唑基、1-異喹啉基、5-異喹啉基、2-喹喔啉基、5-喹喔啉基、3-喹啉基和6-喹啉基。用於以上提到的芳基和雜芳基環系的每一種的取代基選自下文描述的可接受的取代基的組。術語「亞芳基」和「雜亞芳基」分別指芳基和雜芳基的二價形式。為簡潔起見,當與其他術語組合使用時(例如,芳基氧代、芳基硫代、芳基烷基),術語「芳基」包括如以上定義的芳基和雜芳基環二者。因此,術語「芳基烷基」和「雜芳基烷基」意指包括其中芳基或雜芳基被附連至烷基基團的那些基團(例如,苄基、苯乙基、吡啶基甲基、呋喃基甲基等),包括其中碳原子(例如,亞甲基)已被,例如,氧原子(例如,苯氧基甲基、2-吡啶基氧基甲基、3-(1-萘氧基)丙基等)代替的那些烷基。然而,如本文使用的術語「滷代芳基」意指僅包括被一個或更多個滷素取代的芳基。當雜烷基、雜環烷基或雜芳基包括指定數目的成員(例如「3元至7元」)時,術語「成員」指碳或雜原子。如本文使用的,術語「烷基芳基」包括被如以上定義的芳基基團取代的如以上定義的烷基。芳基基團可在烷基基團上的任何點處連接。術語C4-C16烷基芳基包括具有總計4個至16個碳原子的烷基芳基基團,將烷基基團和芳基基團上的碳原子統計在一起。烷基芳基基團的實例包括但不限於苄基(苯基甲基)、苯基乙基和萘基甲基。烷基芳基基團可以是被取代的或未被取代的。只要一個或更多個取代基中的總原子不大於烷基芳基基團,取代基不被計入烷基芳基基團中的原子總數。此外,如本文使用的通常由下式表示的結構:指環結構,例如,但不限於3-碳、4-碳、5-碳、6-碳、7-碳等,脂族和/或芳族環狀化合物,包括飽和環結構、部分飽和環結構和不飽和環結構,包含取代基R基團,其中R基團可以存在或不存在,並且當存在時,一個或更多個R基團可各自在環結構的一個或更多個可用的碳原子上被取代。R基團的存在或不存在和R基團的數目由變量「n」的值確定,變量「n」為通常具有範圍從0至環上可用於取代的碳原子數目的值的整數。如果R基團多於一個,每個R基團,在環結構的可用碳上而不是在另一個R基團上被取代。例如,其中n為0至2的以上結構將包括化合物基團,其包括但不限於:等。代表環狀環結構中的鍵的虛線指示,該鍵可以存在或不存在於環中。即,代表環狀環結構中的鍵的虛線指示,該環結構選自由以下組成的組:飽和環結構、部分飽和環結構和不飽和環結構。帶有斷裂鍵的取代基(諸如下文示出的實例),意指,該取代基與分子在所示位置處直接鍵合。不暗示另外的亞甲基(CH2)基團。符號指示部分與分子的剩餘部分的附連點。帶有兩個斷裂鍵的取代基(諸如下文示出的實例)意指,原子的定向是如所示的,由左至右,並且應該以示出的定向插入到分子中。除非具體說明,不暗示另外的亞甲基(CH2)基團。當芳族環或雜環芳族環的命名原子被定義為「不存在」時,命名原子被直接鍵代替。以上術語(例如,「烷基」、「雜烷基」、「環烷基」和「雜環烷基」、「芳基」、「雜芳基」、「膦酸酯基(phosphonate)」和「磺酸酯基(sulfonate)」及其二價衍生物)的每個意指包括指示基團的被取代和未被取代的兩種形式。下文提供了每種類型的基團的任選的取代基。用於烷基、雜烷基、環烷基、雜環烷基單價和二價衍生基團(包括通常稱為亞烷基、烯基、雜亞烷基、雜烯基、炔基、環烷基、雜環烷基、環烯基和雜環烯基的那些基團)的取代基可以是以數目範圍為從零至(2m』+l)的選自、但不限於以下的多種基團的一種或更多種:-OR』、=O、=NR』、=N-OR』、-NR』R」、-SR』、-滷素、-SiR』R」R』」、-OC(O)R』、-C(O)R』、-CO2R』、-C(O)NR』R」、-OC(O)NR』R」、-NR」C(O)R』、-NR』-C(O)NR」R』」、-NR」C(O)OR』、-NR-C(NR』R」)=NR』」、-S(O)R』、-S(O)2R』、-S(O)2NR』R」、-NRSO2R』、-CN和-NO2,其中m』為此類基團中的碳原子總數。R』、R」、R″'和R""各自可獨立地指氫、被取代的或未被取代的雜烷基、被取代的或未被取代的環烷基、被取代或未被取代的雜環烷基、被取代的或未被取代的芳基(例如,被1-3個滷素取代的芳基)、被取代的或未被取代的烷基、烷氧基或硫代烷氧基基團或芳基烷基基團。如本文使用的「烷氧基」基團為通過二價氧與分子的剩餘部分附連的烷基。當本公開內容的化合物包括多於一個R基團時,例如,當存在多於一個這些基團時,每個R基團獨立地選自R』、R"、R」'和R""基團。當R'和R"被附連到同一氮原子時,它們可與氮原子組合形成4-元、5-元、6-元或7-元環。例如,-NR』R」意指包括,但不限於,1-吡咯烷基和4-嗎啉基。從取代基的以上討論中,本領域技術人員將理解,術語「烷基」意指包括包含與除了氫基團以外的基團鍵合的碳原子的基團,諸如滷代烷基(例如,-CF3和-CH2CF3)和醯基(例如,-C(O)CH3、-C(O)CF3、-C(O)CH2OCH3等)。類似於對於以上烷基基團描述的取代基,用於芳基和雜芳基基團(以及它們的二價衍生物)的示例性取代基是變化的,並且以數目範圍為從零至芳族環系統上開放化合價的總數目選自,例如:滷素、-OR』、-NR』R」、-SR』、-滷素、-SiR』R」R』」、-OC(O)R』、-C(O)R』、-CO2R』、-C(O)NR』R」、-OC(O)NR』R」、-NR」C(O)R』、-NR』-C(O)NR」R』」、-NR」C(O)OR』、-NR-C(NR』R」R』」)=NR」」、-NR-C(NR』R」)=NR』」-S(O)R』、-S(O)2R』、-S(O)2NR』R」、-NRSO2R』、-CN和-NO2、-R』、-N3、-CH(Ph)2、氟代(C1-C4)烷氧代及氟代(C1-C4)烷基;並且其中R'、R"、R″'和R""可獨立地選自氫、被取代的或未被取代的烷基、被取代的或未被取代的雜烷基、被取代的或未被取代的環烷基、被取代的或未被取代的雜環烷基、被取代的或未被取代的芳基和被取代的或未被取代的雜芳基。當本公開內容的化合物包括多於一個R基團時,例如,當存在多於一個這些基團時,每個R基團對每個R』、R"、R″'和R""基團獨立地選擇。芳基或雜芳基環的相鄰原子上的兩個取代基可任選地形成式-T-C(O)-(CRR』)q-U-的環,其中T和U獨立地為-NR-、-O-、-CRR』-或單鍵,且q為從0至3的整數。可選地,芳基或雜芳基環的相鄰原子上的兩個取代基可任選地被式-A-(CH2)r-B-的取代基代替,其中A和B獨立地為-CRR』-、-O-、-NR-、-S-、-S(O)-、-S(O)2-、-S(O)2NR』-或單鍵,且r為從1至4的整數。如此形成的新環的單鍵之一可任選地被雙鍵代替。可選地,芳基或雜芳基環的相鄰原子上的兩個取代基可以任選地被式--(CRR』)s--X』--(C」R』」)d--的取代基代替,其中s和d獨立地為從0至3的整數,且X』為-O-、-NR』-、-S-、-S(O)-、-S(O)2-或-S(O)2NR』-。取代基R、R'、R」和R』」可獨立地選自氫、被取代的或未被取代的烷基、被取代的或未被取代的環烷基、被取代的或未被取代的雜環烷基、被取代的或未被取代的芳基和被取代的或未被取代的雜芳基。如本文使用的術語「醯基」指有機酸基團,其中羧基基團的--OH已經被另一取代基代替並且具有通式RC(=O)--,其中R為如以上定義的烷基、烯基、炔基、芳基、碳環、雜環或芳族雜環基團。因此,術語「醯基」特別地包括芳基醯基,諸如乙醯基呋喃和苯甲醯甲基基團。醯基基團的特定實例包括乙醯基和苯甲醯基。術語「烷氧基(alkoxyl)」或「烷氧基(alkoxy)」在本文中可互換使用,並且指通過氧原子被附連至母體分子部分的飽和(即,烷基-O-)或不飽和(即,烯基-O-和炔基-O-)的基團,其中術語「烷基」、「烯基」和「炔基」如先前描述的,並且可包括包含C1-20的線性、支鏈或環狀的、飽和或不飽和的氧代烴鏈,包括例如,甲氧基、乙氧基、丙氧基、異丙氧基、正丁氧基、仲丁氧基、叔丁氧基和正戊氧基、新戊氧基、正己氧基等。如本文使用的術語「烷氧基烷基」指烷基-O-烷基醚,例如,甲氧基乙基或乙氧基甲基基團。「芳氧基」指芳烷基-O--基團,其中芳基基團如先前描述的,包括被取代的芳基。如本文所用的術語「芳氧基」可指苯基氧基或己基氧基及烷基、被取代的烷基、滷代或烷氧基取代的苯基氧基或己基氧基。「芳烷基」指芳基-烷基-基團,其中芳基和烷基如先前描述的,並且包括被取代的芳基和取代的烷基。示例性的芳烷基包括苄基、苯基乙基和萘基甲基。「芳烷基氧基」指芳烷基-O-基團,其中芳烷基如先前描述的。示例性芳烷基氧基基團是苄氧基。「烷氧基羰基」指烷基-O--CO-基團。示例性的烷氧基羰基基團包括甲氧基羰基、乙氧基羰基、丁氧基羰基和叔丁氧基羰基。「芳氧基羰基」指芳基-O-CO–基團。示例性的芳基氧基羰基基團包括苯氧基-羰基和萘氧基-羰基。「芳烷氧基羰基」指芳烷基-O-CO–基團。示例性芳烷氧基羰基是苄氧基羰基。「氨基甲醯基」指式–CONH2的醯胺基團。「烷基氨基甲醯基」指R』RN–CO–基團,其中R和R'的一個為氫,且R和R'的另一個為如先前描述的烷基和/或被取代的烷基。「二烷基氨基甲醯基」指R』RN–CO–基團,其中R和R'的每個獨立地為如先前描述的烷基和/或被取代的烷基。如本文使用的術語羰基二氧基指式–O—CO—OR的碳酸酯基團。「醯基氧基」指醯基-O-基團,其中醯基如先前描述的。術語「氨基」指–NH2基團,並且還指通過用有機基代替一個或更多個氫基的來源於氨的如本領域已知的含氮基團。例如,術語「醯基氨基」和「烷基氨基」分別指具有醯基和烷基取代基基團的特定的N-取代的有機基。如本文使用的「氨基烷基」指與亞烷基連接基共價鍵合的氨基。更具體地,如本文使用的術語烷基氨基、二烷基氨基和三烷基氨基分別指通過氮原子附連至母體分子部分的一個、兩個或三個如先前定義的烷基基團。術語烷基氨基指具有結構–NHR』的基團,其中R'為如先前定義的烷基;而術語二烷基氨基指具有結構–NR』R」的基團,其中R'和R」各自獨立地選自由烷基基團組成的組。術語三烷基氨基指具有結構–NR』R」R」』的基團,其中R'、R」和R』」各自獨立地選自由烷基基團組成的組。另外,R』、R」和/或R』」一起可任選地為–(CH2)k–,其中k為從2至6的整數。實例包括,但不限於,甲基氨基、二甲基氨基、乙基氨基、二乙基氨基、二乙基氨基羰基、甲基乙基氨基、異丙基氨基、哌啶子基(piperidino)、三甲基氨基和丙基氨基。氨基基團為-NR'R」,其中R'和R」通常選自氫、被取代的或未被取代的烷基、被取代的或未被取代的雜烷基、被取代的或未被取代的環烷基、被取代的或未被取代的雜環烷基、被取代的或未被取代的芳基或者被取代的或未被取代的雜芳基。術語烷基硫醚和硫代烷氧基指通過硫原子附連至母體分子部分的飽和(即,烷基-S-)或不飽和(即,烯基-S-和炔基-S-)基團。硫代烷氧基部分的實例包括,但不限於,甲基硫代、乙基硫代、丙基硫代、異丙基硫代、正丁基硫代等。「醯基氨基」指醯基-NH–基團,其中醯基如先前描述的。「芳醯基氨基」指芳醯基-NH–基團,其中芳醯基如先前描述的。術語「羰基」指–(C=O)–基團。術語「羧基」指–COOH基團。此類基團在本文還被稱為「羧酸」部分。如本文使用的術語「滷代」、「滷化物」或「滷素」指氟代、氯代、溴代和碘代基團。另外,術語諸如「滷代烷基」意指包括單滷代烷基和多滷代烷基。例如,術語「滷代(C1-C4)烷基」意指包括,但不限於,三氟甲基、2,2,2-三氟乙基、4-氯丁基、3-溴丙基等。術語「羥基」指-OH基團。術語「羥基烷基」指被-OH基團取代的烷基基團。術語「巰基」指-SH基團。如本文使用的術語「氧代」意指與碳原子或另一種元素雙鍵鍵合的氧原子。術語「硝基」指–NO2基團。術語「硫代」指本文先前描述的化合物,其中碳或氧原子被硫原子代替。術語「硫酸酯基」指–SO4基團。如本文使用的術語硫代羥基或硫醇指式–SH的基團。術語脲基指式–NH—CO—NH2的脲基團。除非另有明確定義,如本文使用的「取代基基團」包括選自本文定義的以下部分的一個或更多個的官能基團:(A)-OH、-NH2、-SH、-CN、-CF3、-NO2、氧代、滷素、未被取代的烷基、未被取代的雜烷基、未被取代的環烷基、未被取代的雜環烷基、未被取代的芳基、未被取代的雜芳基,以及(B)被選自以下的至少一個取代基取代的烷基、雜烷基、環烷基、雜環烷基、芳基和雜芳基:(i)氧代、-OH、-NH2、-SH、-CN、-CF3、-NO2、滷素、未被取代的烷基、未被取代的雜烷基、未被取代的環烷基、未被取代的雜環烷基、未被取代的芳基、未被取代的雜芳基,以及(ii)被選自以下的至少一個取代基取代的烷基、雜烷基、環烷基、雜環烷基、芳基和雜芳基:(a)氧代、-OH、-NH2、-SH、-CN、-CF3、-NO2、滷素、未被取代的烷基、未被取代的雜烷基、未被取代的環烷基、未被取代的雜環烷基、未被取代的芳基、未被取代的雜芳基,以及(b)被選自以下的至少一個取代基取代的烷基、雜烷基、環烷基、雜環烷基、芳基或雜芳基:氧代、-OH、-NH2、-SH、-CN、-CF3、-NO2、滷素、未被取代的烷基、未被取代的雜烷基、未被取代的環烷基、未被取代的雜環烷基、未被取代的芳基、及未被取代的雜芳基。如本文使用的「低級取代基」或「低級取代基基團」意指選自上文對於「取代基基團」描述的所有取代基的基團,其中每個被取代或未被取代的烷基為被取代或未被取代的C1-C8烷基,每個被取代或未被取代的雜烷基為被取代或未被取代的2元至8元雜烷基,每個被取代或未被取代的環烷基為被取代或未被取代的C5-C7環烷基,且每個被取代或未被取代的雜環烷基為被取代或未被取代的5元至7元雜環烷基。如本文使用的「尺寸受限的取代基」或「尺寸受限的取代基基團」意指選自以上對於「取代基基團」描述的所有取代基的基團,其中每個被取代或未被取代的烷基為被取代或未被取代的C1-C20烷基,每個被取代或未被取代的雜烷基為被取代或未被取代的2元至20元雜烷基,每個被取代或未被取代的環烷基為被取代或未被取代的C4-C8環烷基,且每個被取代或未被取代的雜環烷基為被取代或未被取代的4元至8元雜環烷基。在整個說明書和權利要求書中,給定的化學式或名稱應包括所有互變異構體、同類物(congener)、光學異構體和立體異構體、以及其中此類異構體和混合物存在的外消旋混合物。對本領域技術人員將明顯的是,本公開內容的某些化合物可以以互變異構形式存在,所述化合物的所有此類互變異構形式都在本公開內容的範圍內。如本文使用的術語「互變異構體」指兩種或更多種結構異構體之一,其以平衡狀態存在並且容易從一種異構體形式轉化為另一種異構體形式。除非另有說明,本文描述的結構也意指包括該結構的所有立體化學形式;即,對於每個不對稱中心的R和S構型。因此,本發明化合物的單一立體化學異構體以及對映異構體和非對映異構體混合物在本公開內容的範圍內。本公開內容的某些化合物擁有不對稱碳原子(光學或手性中心)或雙鍵;可在絕對立體化學方面定義的對映異構體、外消旋體、非對映異構體、互變異構體、幾何異構體、立體異構體形式,如(R)-異構體或(S)-異構體,如對於胺基酸的(D)-異構體或(L)-異構體,以及單獨的異構體被包括在本公開內容的範圍內。本公開內容的化合物不包括本領域已知的太不穩定以至不能合成和/或分離的那些化合物。本公開內容意指包括呈外消旋形式和光學純形式的化合物。光學活性(R)-異構體和(S)-異構體或者(D)-異構體和(L)-異構體可使用手性合成子或手性試劑製備,或者使用常規技術拆分。當本文描述的化合物包含烯鍵或其他幾何不對稱中心時,且除非另外指明,意圖是化合物包括E和Z幾何異構體兩者。如何製備旋光性形式是本領域熟知的,諸如通過拆分外消旋形式(外消旋體),通過不對稱合成或通過由旋光性原材料合成。外消旋體的拆分可,例如,通過常規方法完成,常規方法諸如在拆分劑的存在下結晶,或使用,例如手性HPLC柱的層析。烯烴,C=N雙鍵等的許多幾何異構體也可存在於本文描述的化合物中,並且所有此類穩定的異構體都被包括在本公開的主題中。描述了本公開的主題的化合物的順式和反式幾何異構體,並且可作為異構體的混合物或作為分離的異構體形式分離。除非明確地指示特定的立體化學或異構體形式,意指結構的所有手性(對映異構體和非對映異構體)和外消旋形式,以及所有幾何異構形式。本文描述的化合物可具有一個或更多個帶電原子。例如,化合物可以是兩性離子的,但可以是總體中性的。取決於pH和其他因素,其他實施方案可具有一個或更多個帶電基團。在這些實施方案中,化合物可與適合的反荷離子締合。如何製備鹽或交換反荷離子是本領域熟知的。通常,此類鹽可通過以下製備:使這些化合物的游離酸形式與化學計量的量的適當的鹼(諸如Na、Ca、Mg或K的氫氧化物、碳酸鹽、碳酸氫鹽等)反應,或通過使這些化合物的游離鹼形式與化學計量的量的適當的酸反應。此類反應通常在水中或在有機溶劑中或在兩者的混合物中進行。可,例如,通過離子交換技術諸如離子交換層析來改變反荷離子。除非明確指示反荷離子或鹽,意指所有兩性離子、鹽和反荷離子。在某些實施方案中,鹽或反荷離子可以是用於施用至受試者的、藥學上可接受的。藥學上可接受的鹽稍後討論。如本文使用的,「保護基團」為可通過容易可得的試劑選擇性去除的化學取代基,所述容易可得的試劑不攻擊分子中的再生的官能基團或其他官能基團。適合的保護基團是本領域已知的並且繼續在開發。適合的保護基團可見於,例如,Wutz等("Greene'sProtectiveGroupsinOrganicSynthesis,第四版,"Wiley-Interscience,2007)中。如由Wutz等(533-643頁)描述的用於保護羧基基團的保護基團,在某些實施方案中被使用。在一些實施方案中,保護基團可通過用酸處理去除。保護基團的特定實例包括但不限於,苄基、對甲氧基苄基(PMB)、叔丁基(tBu)、甲氧基甲基(MOM)、甲氧基乙氧基甲基(MEM)、甲基硫代甲基(MTM)、四氫吡喃基(THP)、四氫呋喃基(THF)、苄氧基甲基(BOM)、三甲基甲矽烷基(TMS)、三乙基甲矽烷基(TES)、叔丁基二甲基甲矽烷基(TBDMS)和三苯基甲基(三苯甲基,Tr)。本領域技術人員將認識到其中需要保護基團的適當的情況並將能夠選擇用於在特定環境中使用的適當的保護基團。除非另有說明,本文描述的結構也意指包括僅在一個或更多個同位素富集原子的存在方面不同的化合物。例如,除了用氘或氚代替氫,或用13C-或14C-富集的碳代替碳以外,具有本發明結構的化合物在本公開內容的範圍內。本公開內容的化合物還可在構成此類化合物的一個或更多個原子處包含非天然比例的原子同位素。例如,化合物可用放射性同位素放射性標記,放射性同位素諸如例如氚(3H)、碘-125(125I)或碳-14(14C)。本公開內容的化合物的所有同位素變體,無論是否是放射性的,都被包括在本公開內容的範圍內。ii.藥學上的鹽本公開內容的化合物可作為藥學上可接受的鹽存在。取決於本文描述的化合物上發現的特定取代基部分,術語「藥學上可接受的鹽」意指包括用相對無毒的酸或鹼製備的活性化合物的鹽。藥學上可接受的鹽通常是本領域普通技術人員熟知的,並且可包括,例如,但不限於,乙酸鹽、苯磺酸鹽(benzenesulfonate)、苯磺酸鹽(besylate)、苯甲酸鹽、碳酸氫鹽、酒石酸氫鹽、溴化物、依地酸鈣、carnsylate、碳酸鹽、檸檬酸鹽、乙二胺四乙酸鹽、乙二磺酸鹽、依託酸鹽、乙磺酸鹽、富馬酸鹽、葡庚糖酸鹽、葡糖酸鹽、穀氨酸鹽、乙醇醯基胺基苯吡酸鹽、己基間苯二酚鹽、海巴明、氫溴酸鹽、鹽酸鹽、羥基萘甲酸鹽、碘化物、羥乙基磺酸鹽、乳酸鹽、乳糖醛酸鹽、蘋果酸鹽、馬來酸鹽、扁桃酸鹽、甲磺酸鹽、粘酸鹽、萘磺酸鹽、硝酸鹽、撲酸鹽(雙羥萘酸鹽)、泛酸鹽、磷酸鹽/二磷酸鹽、聚半乳糖醛酸鹽、水楊酸鹽、硬脂酸鹽、鹼式乙酸鹽、琥珀酸鹽、硫酸鹽、鞣酸鹽、酒石酸鹽(例如(+)-酒石酸鹽、(-)-酒石酸鹽或其混合物,包括外消旋混合物)或茶氯酸鹽。這些鹽可通過本領域技術人員已知的方法製備。其他藥學上可接受的鹽可見於,例如,Remington:TheScienceandPracticeofPharmacy(第20版)Lippincott,Williams&Wilkins(2000)中。還包括鹼加成鹽,諸如鈉、鉀、鈣、銨、有機氨基或鎂鹽,或類似的鹽。當本公開內容的化合物包含相對鹼性的官能團時,酸加成鹽可通過使此類化合物的中性形式與足量的所期望的酸(純的或在適合的惰性溶劑中)接觸來獲得。可接受的酸加成鹽的實例包括來源於如下無機酸的那些:如鹽酸、氫溴酸、硝酸、碳酸、一氫碳酸(monohydrogencarbonic)、磷酸、一氫磷酸(monohydrogenphosphoric)、二氫磷酸(dihydrogenphosphoric)、硫酸、一氫硫酸(monohydrogensulfuric)、氫碘酸或亞磷酸等,以及來源於如下有機酸的鹽:如乙酸、丙酸、異丁酸、馬來酸、丙二酸、苯甲酸、琥珀酸、辛二酸、富馬酸、乳酸、扁桃酸、鄰苯二甲酸、苯磺酸、對甲苯磺酸(p-tolylsulfonic)、檸檬酸、酒石酸、甲基磺酸等。還包括了胺基酸諸如精氨酸等的鹽,以及有機酸如葡糖醛酸或半乳糖醛酸等的鹽,參見,例如,Berge等,「PharmaceuticalSalts」,JournalofPharmaceuticalScience,1977,66,1-19)。本公開內容的某些特定的化合物包含鹼性官能團和酸性官能團兩者,其允許化合物被轉化為鹼加成鹽或酸加成鹽。化合物的中性形式可通過使鹽與鹼或酸接觸並以常規方式分離母體化合物來再生。化合物的母體形式在某些物理性質(諸如在極性溶劑中的溶解度)方面不同於多種鹽形式。本公開內容的某些化合物可以以非溶劑化形式以及溶劑化形式(包括水合形式)存在。通常,溶劑化形式等價於非溶劑化形式,並且被包括在本公開內容的範圍內。本公開內容的某些化合物可以以多種結晶形式或無定形形式存在。通常,所有物理形式對於本公開內容所包括的用途是等同的,並且旨在在本公開內容的範圍內。iii.一般定義儘管本文中採用了特定術語,但它們僅用於一般的和描述性的意義,而非用於限制的目的。為了清楚起見,本文提供了具體定義。除非另有定義,本文使用的所有技術術語和科學術語具有與由本發明描述的主題所屬領域的普通技術人員通常理解的相同的含義。在動物中的「癌症」指擁有致癌細胞的典型特徵的細胞的存在,致癌細胞的典型特徵例如,不受控制的增殖、專門化功能的損失、永生、顯著轉移潛能、抗凋亡活性的顯著增加、快速生長和增殖速率以及某些特徵形態和細胞標誌物。在一些情況下,癌細胞將呈腫瘤的形式;此類細胞可局部存在於動物內,或在血流中循環作為獨立的細胞。「對照」意指標準或參考條件。「疾病」意指損害或幹擾細胞、組織、器官、生物體或受試者的正常機能的任何狀況或紊亂。試劑的「有效量」指當與對照相比時足以引起期望生物響應或可測量的差異的試劑的量。如本領域普通技術人員將理解的,有效治療疾病、紊亂、狀況或損傷的特定試劑的絕對量可根據以下此類因素而變化:如待遞送的試劑,施用的方式,受試者的年齡、體重和一般健康狀況,期望的生物學終點,期望的治療效應等。最終,主治臨床醫師將決定適當的量和劑量方案。例如,試劑的「有效量」可以是當化合物用於成像時足以產生可測量的圖像的量,或當化合物用於療法時足以改善疾病的症狀的量。本領域普通技術人員將進一步理解,試劑的有效量可以以單劑量施用,或者可以通過施用多劑量實現。如本文使用的術語「施用」指使受試者與本公開的試劑接觸。根據長期以來的專利法慣例,在該申請包括權利要求書中當使用術語「一(a)」「一(an)」和「該(the)」時,指「一個或更多個」。因此,例如,提及「受試者(asubject)」包括多個受試者,除非上下文清楚地是意思相反的(例如,多個受試者)等等。貫穿該說明書和權利要求書,術語「包含(comprise)」、「包含(comprises)」和「包含(comprising)」被用於在非排他性含義,除非當上下文另有要求時。同樣地,術語「包括(include)」和其語法變體旨在非限制,使得列表中項目的列舉不排除可被取代或添加至所列項目的其他類似的項目。為了該說明書以及所附的權利要求書的目的,除非另有說明,在所有的實例中,在說明書和權利要求書中使用的所有表示量、尺寸、維度、比例、形狀、製劑、參數、百分比、參數、數量、特徵、及其他數值的數字要理解為由術語「約」修飾,即使術語「約」可能未明確地與該值、量或範圍一起出現。因此,除非相反地指示,在以下說明書和所附的權利要求書中列出的數值參數不是精確的且不需要是精確的,但是如所期望的可以是接近的和/或更大或更小,反映了公差、轉換因子、四捨五入、測量誤差等,以及本領域技術人員已知的其他因素,取決於旨在通過本公開的主題獲得的期望特性。例如,當術語「約」指值時能夠用意在於包括與特定的量在一些實施方案中±100%、在一些實施方案中±50%、在一些實施方案中±20%、在一些實施方案中±10%、在一些實施方案中±5%、在一些實施方案中±1%、在一些實施方案中±0.5%、以及在一些實施方案中±0.1%的差異,因為此類差異對於進行所公開的方法或採用所公開的組合物是適當的。此外,當與一個或更多個數值或數值範圍關聯使用時,術語「約」應理解為指所有此類數值,包括範圍內的所有數值以及通過將邊界延伸至高於和低於所列出的數值修改該範圍。通過端點引述的數值範圍包括歸入該範圍內的所有數值例如全部的整數,包括其分數(例如,1至5的引述包括1、2、3、4和5,以及其分數,例如1.5、2.25、3.75、4.1等)以及在該範圍內的任何範圍。實施例以下實施例已被包括以提供本領域普通技術人員用於實踐本發明公開的主題的代表性實施方案的指導。根據本公開內容和本領域技術人員的一般水平,本領域技術人員可以理解,以下實施例預期僅是示例性的,且可採用許多變化、修改和改變而不偏離本公開的主題的範圍。以下合成的描述和具體實施例僅預期用於說明的目的,並且不被解釋為以任何方式限制通過其他方法製備本公開內容的化合物。實施例1合成和評價基於釓(Gd)的造影劑概述磁共振(MR)成像由於可同時提供解剖、功能和分子信息是有利的。MR分子成像可將這種已確立的臨床模式及其高空間解析度的普遍存在與體內分子譜系分析相結合。然而,由於MR的固有的低靈敏度,需要高的局部濃度的生物靶以產生可辨別的MR對比。由於前列腺特異性膜抗原(PSMA)在靶細胞內的高濃度、在非靶組織中的有限表達和細胞表面上的可接近性,我們假設前列腺特異性膜抗原(PSMA),用於前列腺癌的成像和療法的有吸引力的靶,可作為基於MR的分子成像的適合的生物標誌物。出於該目的,已合成了分別用每分子一個至三個Gd-螯合物分級的三種高親和力、低分子量的基於釓(Gd)(III)-的PSMA靶向造影劑Gd1、Gd2和Gd3(圖1A)。本研究的目的是評價合成試劑的PSMA結合親和力和縱向弛豫度(r1)。使用電感耦合等離子體質譜(ICP-MS)評價表達PSMA的同基因細胞、不表達的對照細胞中的試劑的細胞攝取。最後,通過MR成像在體外和體內評價試劑區分表達PSMA的細胞與對照細胞的能力。材料和方法(21S,25S)-8,15,23-三氧代-1-((4-((1,4,7,10-四(羧甲基)-1,4,7,10-四氮雜環十二烷-2-基)甲基)苯基)氨基)-1-硫氧代-2,7,16,22,24-五氮雜二十七烷-21,25,27-三羧酸,Gd1。遵循最近的報導製備化合物Gd1。化合物1如下文描述以三個步驟進行製備。將商購可得的N-Boc-1,4二氨基丁烷(在0.5mlDMSO中的68mg,0.36mmol)與1,4,7,10-四氮雜環十二烷-1,4,7,10-四乙酸、2-[(4-異硫氰酸苯基)甲基](p-SCN-Bn-DOTA)(在2.5mLDMSO中的192mg,0.28mmol)和DIEA(132μl,0.75mmol)混合併在40℃攪拌4h。將溶劑蒸發,並將固體剩餘物使用水和乙腈(在各自中的0.1%TFA)通過反相C18快速層析(5.5g,AgilentSF10)純化,以在凍幹之後獲得Boc-保護的7。產率:146mg,~55%。ESI-MS740[M+H]+。然後將由該步驟得到的化合物用冰冷的TFA/CH2Cl2(1/1)溶液處理,並在環境溫度下放置攪拌2小時。將溶劑蒸發,並將剩餘物在真空下乾燥並通過反相快速層析(5.5g,AgilentSF10)純化,以中等產率產生7。將溶劑蒸發,將剩餘物在真空下乾燥並通過反相快速層析(5.5g,AgilentSF10)純化,以產生7。產率~104mg,40%。1HNMR(DMSO-d6)δ:8.80-8.64(m,1H)、8.12-7.90(m,2H)、7.75-7.10(bm,4H)、4.65-4.63(m,1H)、4.17-2.59(m,27H)、2.40-1.11(m,6H)。ESI-MS:640[M+1]+。將7(在3mL蒸餾水中的110mg,0.17mmol)的溶液添加至溶液Gd2(CO3)3(85mg,0.17mmol)並在60℃下放置攪拌14h。ESI-MS:對於C48H77N10O17S,797.5183[M+H]+的計算值得出為:797.5212。化合物然後通過HPLC純化。方法1:溶劑A(在水中的0.1%TFA)和溶劑B(在乙腈溶液的0.1%TFA),流速為8mL/min。洗脫梯度為100%A和0%B持續5min,且100%A至80%A和0%B至20%B歷時5–25min,且80%A至20%A和20%B至80%B從25-30min。(30S,34S)-2,9,17,24,32-五氧代-1-(4,7,10-三(羧甲基)-1,4,7,10-四氮雜環十二烷-1-基)-8-(2-(4,7,10-三(羧甲基)-1,4,7,10-四氮雜環十二烷-1-基)乙醯氨基)-3,10,16,25,31,33-六氮雜三十六烷-11,30,34,36-四羧酸,二釓(III)鹽。Gd2。化合物遵循方案2製備。向N-Bis-Boc-L-賴氨酸NHS(3gm,在10mLDMF中的6.7mmol)的溶液添加Fmoc-Lys(Boc)-OH(2.49g,6.7mmol),並將溶液在rt超聲1小時直至獲得澄清溶液。將溶液在rt攪拌4h,並在真空下去除溶劑,以幾乎定量的產率獲得4。將化合物4使用3/97MeOH/CH2Cl2作為洗脫液通過矽膠柱進一步純化。1HNMR(CDCl3)δ:8.01(d,2H)、7.89(m,2H)、7.78-7.44(m,4H)、6.82(m,1H)、6.15(m,1H)、5.58(m,1H)、5.01-4.03(m,5H)、3.75-3.32(m,6H)、2.22-1.31(m,30H)。ESMSm/Z:696[M+H]+。將化合物4(2g,2.9mmol)溶解於10mL1/1TFA/CH2Cl2溶液中,並在rt放置攪拌2h。在溶劑蒸發之後,將固體剩餘物用3××3mL乙醚洗滌,並在真空下乾燥,以產生5,作為TFA鹽。化合物5以定量產率獲得,並在凍幹之後使用而不經進一步純化。1HNMR(D2O)δ:8.01(d,2H)、7.89(m,2H)、7.78-7.44(m,4H)、4.78-4.75(m,2H)、4.32(m,1H)、4.11-4.09(m,1H)、4.01-3.98(t,1H),3.50-3.11(m,3H)、3.10-2.99(m,2H)、2.01-1.01(m,12H)。在rt,經45min的時間段,向DOTA-NHS(在0.5mLDMSO中的100mg,0.13mmol)的溶液以小份添加5.2TFA(32mg,0.04mmol)和DIEA(0.78mmol,136μL)。然後將溶液攪拌持續另一2h,且反應的完成使用HPLC監測。在反應完成之後,反應通過HPLC純化,以獲得6。將化合物6用20%哌啶溶液處理至Fmoc基團,並使用90/10H2O/CH3CN(在每個中的0.1%TFA)溶液使用C18快速層析純化並凍幹。ESIMS:1046[M+H]+。將該凍幹的化合物(50mg,0.047mmol)溶解於蒸餾水(2mL)中,添加至Gd2(CO3)3(在3mL水中的0.26mmol)的溶液中,並在60℃攪拌12h。化合物7使用90/10至80/20H2O/CH3CN(在每個中的0.1%TFA)溶液的梯度,使用C18快速層析純化並凍幹。向在DMSO中的3(25mg,0.004mmol)的溶液緩慢添加7(40mg,0.003mmol)持續30min,並在rt攪拌約2h直至反應完成。反應的完成使用HPLC監測。在完成之後,反應混合物隨後通過HPLC純化,並將產物凍幹。ESI-MS:1813.08[M+H]+,得出為:1813.08。化合物然後通過HPLC純化。方法1:對於C64H103Gd2N15O26,1813.5681[M]+的計算值得出為1813.5681[M+1]。溶劑A(在水中的0.1%TFA)和溶劑B(在乙腈中的0.1%TFA),流速為8mL/min。洗脫梯度為100%A和0%B持續5min,且100%A至80%A和0%B至20%B歷時5–25min,且80%A至20%A和20%B至80%B從25-30min。(3S,7S)-5,13,20,28-四氧代-32-(2,4,6-三(1-(2-羥基-3-(4,7,10-三(羧甲基)-1,4,7,10-四氮雜環十二烷-1-基)丙基)-1H-1,2,3-三唑-4-基)苯氧基)-4,6,12,21,27-五氮雜三十二烷-1,3,7,22-四羧酸,三釓(III)鹽。Gd3。Gd3通過使用如方案3中示出的多步合成來製備。遵循先前的報導製備化合物8。2,5-二氧代吡咯烷-1-基5-(2,4,6-三乙炔基苯氧基)戊酸酯,9。向8(在5mLDMF中的300mg,1.13mmol)的溶液添加TSTU(440mg,1.47mmol)和TEA(541μL,3.39mmol),並且將所得的溶液在室溫放置攪拌4h,直至反應通過TLC監測完成。在高真空下去除溶劑,將剩餘物溶解於CH2Cl2中,並使用40/60至50/50EtOAc/己烷溶液作為洗脫液通過矽膠柱純化。將包含產物的級分合併在一起並蒸發,以獲得為無色固體的期望的產物。產率~310mg。NMR(CDCl3):δ7.56(s,2H)、4.26(t,2H)、3.39(s,2H)、3.04(s,1H)、2.78(s,4H)、2.48(t,2H)、2.01-1.80(m,4H)。(3S,7S)-26-氨基-5,13,20-三氧代-4,6,12,21-四氮雜二十六烷-1,3,7,22-四羧酸2,2,2-三氟乙酸鹽,10。遵循先前的報導製備化合物10。簡言之,向Tris-t-Bu保護的3(在1.35mlDMF中的100mg,0.135mmol)的溶液添加H-Lys(Boc)(O-t-Bu)(59.5mg,0.175mmol),隨後是DIEA(70.7μL,0.135),並將澄清溶液在rt攪拌過夜。然後將溶液在真空下濃縮至澄清的油狀剩餘物。將剩餘物溶解於2:1的MeCN/水(6mL)中並凍幹,以獲得澄清泡沫狀產物產率。使用產物而不經進一步純化。產率:117mg,0.126mmol,93%。ESI-MS:928[M+H]+。將化合物溶解於冰冷的2mlTFA/CH2Cl2的溶液中,隨後逐滴添加TES(278μL,1.7mmol)。將澄清溶液保持攪拌5h,在真空下濃縮。將剩餘物溶解於5mL水中,並使用反相快速層析純化。產物使用80/20水/CH3CN(在每個中的0.1%TFA)洗脫。ESI-MS:603[M+H]+。(3S,7S)-5,13,20,28-四氧代-32-(2,4,6-三乙炔基苯氧基)-4,6,12,21,27-五氮雜三十二烷-1,3,7,22-四羧酸,11。將化合物9(132mg,0.362mmol)一次性添加至包含以下的溶液:(3S,7S)-26-氨基-5,13,20-三氧代-4,6,12,21-四氮雜二十六烷-1,3,7,22-四氮雜羧酸2,2,2-三氟乙酸鹽(260mg,0.362mmol)、三乙胺(0.202mL,1.44mmol)和DMF(3.62mL)。將混合物在室溫攪拌4h,並濃縮至棕褐色剩餘物。將剩餘物溶解於1/1水/乙腈(3mL)中,並使用C18反相快速層析純化,其中步驟梯度由100%水、0.1%TFA,隨後是80/20,60/40水/乙腈(在每個中的0.1%)組成。每個梯度步驟由約144mL溶劑體積組成。流速為40mL/min。將包含期望產物的級分濃縮至剩餘物並凍幹,以產生為白色固體的(3S,7S)-5,13,20,28-四氧代-32-(2,4,6-三乙炔基苯氧基)-4,6,12,21,27-五氮雜三十二烷-1,3,7,22-四羧酸。169mg,54%產率。ESI-MS,對於C43H57N5O13[M+H]+852.4的計算值,得出為851.9。1HNMR(400MHz,DMSO-d6)12.12(bs,4H)8.01(d,1H)、7.76(m,2H)、7.57(s,2H)、6.33(m,2H)、4.47(s,2H)、4.28(s,1H)、4.09-4.15(m,4H)、3.00(m,4H)、2.21-2.27(m,2H)、2.10(m,4H)、2.02(t,2H)、1.89-1.94(m,1H)、1.22–1.69(m,24H)。13CNMR(100MHz,DMSO-d6)δ175.0、174.6、174.3、174.1、172.8、172.3、172.1、162.1、158.9、158.5、157.7、137.6、118.0、117.5、86.6、82.0、81.5、78.6、74.1、52.7、52.1、38.7、38.6、35.8、35.5、32.2、31.1、30.3、29.7、29.3、29.2、28.9、28.8、27.9、25.7、25.6、23.3、23.0、22.1。Gd3。向包含(3S,7S)-5,13,20,28-四氧代-32-(2,4,6-三乙炔基苯氧基)-4,6,12,21,27五氮雜三十二烷-1,3,7,22-四羧酸(12mg,0.14mmol)、化合物002(28mg,0.046mmol)和叔丁醇(0.1mL)的混合物添加水(0.05mL),隨後是TBTA(0.15mg,0.3μmol)和六氟磷酸四乙腈銅(I)(tetrakis(acetonitrile)copper(I)hexafluorophosphate)(0.11mg,0.3μmol)。將混合物在65℃攪拌18h。將反應混合物溶解於2.5mL的0.1%碳酸氫鈉中並過濾。因此獲得的溶液使用Phenomenex,Luna,10微米,10×250mm柱和由0-95%乙腈:水組成的梯度在HPLC上經20分鐘純化。將期望的產物(003)在6.1-7.1分鐘時洗脫。將包含003的級分合併,濃縮並凍幹,以得到白色固體。13mg,34%產率。ESI-MS,對於C94H141Gd3N26O34[M-H]-2650.7的計算值,得出為2648.9。方案1方案2方案3結果代表性的PSMA造影劑單體-、二聚體-和三聚體Gd(Gd1、Gd2和Gd3)的結構示於圖1A中,其包含Lys-Glu脲作為靶向部分。開發了多步溶液相合成方法以製備靶化合物,並且在方案1-3中概述。由於DOTA形成具有高熱力學和動力學穩定性的絡合物,對於所有三種化合物,使用螯合劑DOTA。Gd1包含DOTA-Bn-SCN以提供更高的弛豫度。Gd1的結構是基於最近報導的用於正電子發射斷層攝影術(PET)的鉛86Y標記的成像劑,其在臨床前模型中顯示出高的和特異性的腫瘤累積(Banerjee,等2015)。Gd2通過採用基於溶液的肽合成策略將賴氨酸的∝-胺和ε-胺二者與DOTA-NHS綴合來製備。在相同條件下,當反應在室溫在超聲浴中進行時,偶聯反應的產率顯著改進。Gd3包含酚核,該酚核與三個Gd(III)-DOTA使用點擊化學反應(clickchemistry)通過剛性三唑鍵合結合,如由Mastarone等先前報導的。將靶向PSMA的官能團通過酚氧綴合至該核。Gd3由於三唑連接基部分的剛性增加而顯示出相對高的弛豫度。化合物通過反向HPLC純化並通過LCMS表徵。為了確定試劑的包含Gd(III)的部分對探針的結合親和力的任何潛在負效應,對於Gd1、Gd2和Gd3的PSMA抑制常數(Ki)值使用基於螢光的PSMA抑制測定確定並列於表1中。表1.造影劑的物理性質[a]列出的弛豫度分別指示試劑的離子/分子弛豫度。[b]ZJ43(Ki0.29;Ki的95%CI0.22-0.39nM)。使用已知的高親和力PSMA抑制物,N-[[[(S)-1-羧基-3-甲基丁基]氨基]羰基]-L-穀氨酸(ZJ43)(Olszewski等,2004)作為參考配體。如預期的,所有化合物顯示出高的結合親和力,其中Ki值範圍是Gd1的最高(0.45nM),隨後是Gd3(7.19nM)且Gd2(18.18nM)最低。當在9.4T和25℃下成像時,溶液幻影(solutionphantom)指示在PBS中的r1弛豫度在每Gd(III)3.0和6.2mM-1s-1之間以及每造影劑3.0和12.5mM-1s-1之間變化(表1)。如預期的,在25℃,Gd1具有最低的弛豫度,隨後是Gd2和Gd3。為了確定試劑的選擇性和特異性,選擇經遺傳修飾以表達高量PSMA的人類前列腺癌細胞(PC3PIP)和作為陰性對照的對應的野生型未表達PSMA的細胞(PC3flu)(Banerjee,Angew.,2001)。與Gd1或Gd2孵育之後,沉澱的PSMA+PC3PIP和PSMA-PC3flu細胞未顯示出T1-加權的MR對比或R1的變化。相反,與未標記的細胞以及與PSMA-PC3flu細胞沉澱相比,與Gd3一起孵育的兩種細胞系的T1-加權圖像顯示出顯著的MR對比增強,如圖4A中示出的。通過在50μMGd3的存在和不存在下的細胞沉澱的MRI的增強和T1測量示出了,在通過用標準介質洗滌從細胞系統去除Gd3之後,與Gd3-處理的flu細胞和對照flu對照細胞相比,較高的增強和在Gd3處理的PIP細胞和對照PIP細胞之間的T1弛豫率差異ΔR1(圖4B和圖4C)。通過進行Gd3和ZJ43的共孵育的選擇性阻斷實驗確實顯示T1增強的顯著阻斷。在ZJ43存在下與Gd3孵育的細胞在兩種類型的細胞中僅顯示T1值的微小變化,指示,ZJ43能夠特異性阻斷Gd3的結合。這些結果指示,Gd3表現出對PSMA+PC3PIP細胞的受體特異性細胞結合,並表現出PSMA-介導的對比增強,證明受體介導的胞吞作用的概念。在成像分析之後,對細胞的ICP-MS分析確實顯示存在與PC3flu細胞沉澱締合的可忽略不計的Gd(III),而PC3PIP細胞沉澱具有高的Gd量(圖2和圖3)。PSMA+PC3PIP細胞沉澱對於Gd3具有~22.82μM的估計的細胞內Gd(III)濃度,隨後是對於Gd2和Gd1分別為~12.5μM和~7.2μM的估計的細胞內Gd(III)濃度(圖2)。因此,PIP細胞和flu細胞的R1之間的差異反映了由於PIP細胞中特異性Gd3與PSMA結合而引起的變化。細胞內化測定顯示,在PSMA+PC3PIP細胞中對於Gd1和Gd2經歷內化的孵育劑量的百分比(%ID)在孵育4h之後分別為9.06±0.31%ID和21.63±3.51%ID,而那時僅有2.42±0.11%ID和3.51±1.32%ID與細胞表面締合(圖3)。此外,稍高的非特異性攝取與PSMA-PC3flu細胞中的Gd2相關,這可能與該試劑與Gd1相比的較低的Ki值相關。為了進一步檢查細胞攝取和內化,製備用羅丹明-RedTM-X標記的雙模式Gd單體造影劑,以確認PSMA-介導的這類造影劑的內化(圖5D)。如預期的,該試劑僅在PSMA+PC3PIP細胞中顯示出特異性和高累積(圖5A至圖5C)。這些結果顯示,在該水平表達的細胞受體可使用這些簡單的靶向劑通過MRI檢測。在孵育1h、4h和24h之後,對於Gd3進行時間依賴性內化研究(圖6A和圖6B)。在PSMA+PC3PIP細胞中,在1h和4h時的細胞內攝取是高的和特異性的,分別為28.30±0.47和39.92±3.59%ID,而在孵育後24h時,觀察到~89.69±3.90%ID。在那些相同時間點,相似量的Gd(~33%-37%ID)與細胞膜締合。這些結果表明,在PSMA+PC3PIP細胞沉澱中Gd3的可檢測的T1-加權增強與在PSMA+PIP細胞中Gd3的高的特異性累積充分相關。在評價Gd3用於活小鼠成像之前,Gd3的生物相容性使用細胞增殖測定檢查。將多種濃度的Gd3與PSMA+PC3PIP和PSMA-PC3flu細胞孵育24小時。多達1mM的Gd(III)濃度的Gd3對PSMA-flu細胞的存活力不具有顯著效應(即~90%存活力)(圖7)。然而,>1mM的Gd(III)濃度影響PSMA+PC3PIP細胞存活力(圖8)。觀察到的PSMA+PC3PIP細胞死亡的水平可歸因於Gd3的高的細胞內化,以及採用的長孵育時間(24h)。體內MR成像通過使用攜帶分別在右下脅腹和左下脅腹皮下植入的PSMA+PC3PIP(右側)和PSMA-PC3flu(左側)腫瘤的雄性NOD/SCID小鼠以9.4T靜脈注射Gd3(0.05mmol/Kg劑量)之後進行。在注射之後的前20min期間,PIP和flu腫瘤接受非特異性攝取。在所有組織中觀察到T1值的急劇下降,PSMAflu最高0.63秒,然後是PIP腫瘤0.57秒和肌肉0.261秒。顯著地,從肌肉和flu腫瘤觀察到造影劑的快速清除。在注射後40min時,PIP腫瘤處的對比增強最高,為36%,並且直至注射後1.5h保持高達30%。在3h時,發現肌肉和flu腫瘤的T1值回到初始值,而PIP腫瘤的T1值未顯示顯著變化。Gd3的體內MR成像還對攜帶分別在右下脅腹和左下脅腹皮下植入的PSMA+PC3PIP和PSMA-PC3flu腫瘤異種移植物的小鼠在單次快速推注式靜脈注射(0.06mmol/kg)之後進行。圖9A顯示注射後40min至160min時的兩個腫瘤的1mm切片的定量對比增強映射(ΔR1)。對比增強在PSMA+PC3PIP腫瘤內保持恆定持續至少3h,但在PSMA-PC3flu腫瘤和肌肉組織內迅速降低。在前40min至60min內,PSMA+PC3PIP腫瘤(圖10A和圖10B)的T1值的變化達到最小值1,819±76ms(平均值±SD,R1值的平均36%增強,n=4),並以29%保持恆定直至90min,並在注射之後在190min時緩慢降低至24%。對於PSMA-PC3flu腫瘤,在注射後20min時最高對比增強為~24%,隨後對比增強快速衰減(在40min之後ΔR1<20%)。這些結果顯示在注射後80min和120min時對於PSMA+PC3PIP腫瘤的特異性對比增強(P≤0.05)。如圖9B中示出的,將這些結果與使用無靶向部分的三聚體Gd-探針以相同方式給藥的其他小鼠直接比較,這未顯示腫瘤增強(Mastarone,2011)。在相同的實驗條件下,用鹽水(PBS)的對照研究未顯示PIP和flu腫瘤的T1值的任何變化(圖12)。不希望受任何一種特定理論所束縛,在與PSMA結合後,Gd3的旋轉相關時間(rotationalcorrelationtime)相對於未結合狀態增加。結合還可改變對於每種試劑的水合數目和水交換速率,這可改變弛豫度值不同於從游離造影劑的弛豫度預期的那些值(Caravan等,2007)。此外,在高場,由於造影劑與細胞組分的相互作用,增加旋轉相關時間可稍微降低弛豫度(Caravan,P.,等2009;DeLeon-Rodriguez,L.M.,等2010;Geninatti-Crich,S.2011)。通過利用靈敏的多聚體Gd(III)複合物與已確立的靶向PSMA的小分子組合,進行體外和體內PSMA靶向的MR分子成像。總之,已顯示,基於Gd的造影劑Gd3可用於使用小鼠異種移植物的體內PSMA特異性MR成像。描述的用於前列腺和其他癌症中的平移使用的構建體的優化正在進行中。實施例2PSMA的受體濃度每細胞受體數(N.R.C.)=4.9x106個,r細胞=8.75μm因此,為了觀察到0.05秒-1的變化(約10%增強,考慮到組織T01=2秒),需要弛豫度(如果受體:對比1:1)實施例386Y-標記的前列腺特異性膜抗原抑制物的臨床前評價用於劑量學估計概述86Y(半衰期=14.74h,33%β+)在具有相對長的物理半衰期的新興類型的發射正電子的同位素內,使得能夠擴展生物過程的成像。已報導了在齧齒動物實驗模型中三種低分子量86Y-標記的PSMA結合脲的製備和生物分布的研究(圖14),以及在非人類靈長類動物中的藥物動力學上最有利的試劑用於在製備用對應的90Y-和177Lu-標記的試劑的臨床試驗中的放射劑量學的成像。在製備[86Y]-4-6中使用多步合成。PSMA抑制常數通過競爭性結合測定評價。使用攜帶腫瘤的雄性小鼠的體內表徵直到注射之後24h通過對於[86Y]-4-6的PET/CT和通過[86Y]-4和[86Y]-6的生物分布研究進行。記錄定量全身PET掃描以測量使用[86Y]-6的雄性狒狒中的14個器官的動力學。化合物[86Y]-4-6以高放射化學產率和純度獲得,具有大於83.92GBq/μmol的比放射性。使用攜帶PSMA1/2陽性PC-3PIP和PSMA-陰性PC-3flu腫瘤的小鼠的PET成像和生物分布研究揭示,[86Y]-4-6在注射之後20min時開始在PSMA陽性PC-3PIP腫瘤中具有高位點特異性攝取並且在24h時仍保持高的。化合物[86Y]-6顯示最高的腫瘤攝取和保留,在5h和24h時分別具有每克32.17±7.99和15.79±6.44百分比注射劑量(%ID/g)。低活性濃度與血液和正常器官(除了表達PSMA的組織腎以外)相關。在狒狒中的PET成像顯示,所有器官具有2相(快速和緩慢)清除率,在25min時在腎中具有最高攝取(8%ID/g)。使用單獨的絕對攝取動力學來使用OLINDA/EXM軟體計算放射劑量。最高的平均吸收劑量由腎皮質接收,具有1.9mGy/[86Y]-6的MBq。材料和方法從商業來源獲得的溶劑和化學品是分析級或更好的,且在沒有進一步純化的情況下使用。所有9-芴基甲氧基羰基(Fmoc)保護的胺基酸,包括Fmoc-Lys(Boc)-Wang樹脂、1-羥基苯並三唑一水合物和2-(1H-苯並三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸鹽(HBTU)購自ChemImpexInternationalInc.(Wooddale,IL)。無載體的[86Y](NO3)3從美國國立衛生研究院的國家癌症研究所(theNationalCancerInstituteoftheNationalInstitutesofHealth)(Bethesda)獲得。DOTA-三(叔丁基酯)-單酸和p-SCN-Bn-DOTA(B-205)購自Macrocyclics,Inc.(Dallas,TX)。硝酸釔(III)、三乙基矽烷(Et3SiH)、二異丙基乙胺(DIEA)和三乙胺(TEA)購自Sigma-Aldrich(SaintLouis,MO,USA)。除非另有說明,所有其他化學品購自ThermoFisherScientific(Pittsburgh,PA)。分析薄層層析(TLC)使用Aldrich鋁背襯的0.2mm矽膠Z19,329-1板進行,並通過紫外光(254nm)、I2和在EtOH中的1%茚三酮可視化。快速層析使用購自Bodman(Aston,PA)的矽膠MPSiliTech32-63D進行。所有實驗以一式兩份或一式三份進行以確保再現性。1HNMR譜在BrukerUltrashieldTM400MHz光譜儀上記錄。通過參考由NMR溶劑的不完全氘化產生的質子共振,以低場ppm記錄化學位移(δ)。低解析度ESI質譜在BrukerDaltonicsEsquire3000Plus光譜儀,Billerica,MA上獲得。高分辨質譜通過在BrukermicrOTOF-II上直接注入或通過經由具有用BrukermicrOTOF-QII偶聯的C18柱的超高壓DionexRSLC的LC洗脫使用ESI由theUniversityofNotreDameMassSpectrometry&ProteomicsFacility,NotreDame,IN獲得。4-6和[89Y]4-6的高效液相色譜(HPLC)純化在Waters600EDeltaLC系統與Waters486可用的波長UV/Vis檢測器(兩者通過Empower軟體控制)(WatersCorporation,Milford,MA)上使用PhenomenexC18Luna10××250mm2柱進行(圖15A和圖15B、圖16A和圖16B、以及圖17A、圖17B和圖17C)。HPLC使用溶劑A(在水中的0.1%TFA)和溶劑B(在乙腈中的0.1%TFA)使用以下方法進行。方法1:洗脫梯度為75%A和25%B持續5min,且75%A至60%A和25%B至40%B歷時5–25min,且60%A至75%A和40%B至25%B從25-30min,流速為8mL/min。方法2:流速為8mL/min。洗脫梯度為100%A和0%B持續0-5min,且100%A至45%A和0%B至55%B持續5–45min。[86Y]4-6的HPLC純化在裝備有模型490UV吸光度檢測器和與BioscanFlow-count系統連接的BioscanNaI閃爍檢測器(Bioscan,WashingtonD.C.,USA)的VarianProstar系統(PaloAlto,CA)上進行。對於[86Y]4-6的HPLC純化,使用WatersNovapakC18150x3.9mm2柱。HPLC使用溶劑A(在水中的0.1%TFA)和溶劑B(在CH3CN中的0.1%TFA)和1mL/min的流速使用以下方法進行。使用等度方法(isocraticmethod)85%A和15%B持續25min,以用於純化[86Y]4。採用梯度方法0-5min78%A和22%B,5-25min78%A至58%A和22%B至42%B,以用於[86Y]5。使用梯度方法0-5min88%A和12%B,5-25min88%A至68%A和12%B至32%B,以用於純化[86Y]6。比放射性被計算為在製備HPLC純化期間產物的保留時間時洗脫的放射性除以對應於UV吸收曲線下面積的質量。如通過HPLC確定的,所有最終化合物以>95%放射化學純度獲得。化合物1遵循先前報導(Banerjee,Pullambhatla,Byun,等,2011)製備。化合物4和5通過如早前對於4報導的相同一般方法(Banerjee等,2010)進行製備,且對於5下文簡要提及。合成和放射化學:(13S,27S,31S)-4,7,10-三苄基-2,5,8,11,18,21,29-七氧代-1-(4,7,10-三(羧甲基)-1,4,7,10-四氮雜環十二烷-1-基)-3,6,9,12,17,22,28,30-八氮雜三十三烷-13,27,31,33-四羧酸,5。化合物5遵循先前報導((Banerjee等,2010)製備,如方案4中概述的。化合物3和4通過遵循固相肽策略製備。允許Fmoc-Lys(Boc)-Wang樹脂(100mg,0.43mM)用CH2Cl2(3mL)隨後是DMF(3mL)膨脹。將在DMF中的20%哌啶的溶液(3x3mL)添加至樹脂,其然後在環境溫度在機械振蕩器上輕輕震搖30min。將樹脂用DMF(3x3mL)和CH2Cl2(3x3mL)洗滌。游離胺的形成通過Kaiser測試(Kaiser等,1970)評價。在DMF中膨脹樹脂之後,添加在DMF中的Fmoc-Phe-OH(3當量)、HBTU(3當量)、HOBt(3當量)和DIPEA(4.0當量)的溶液,並輕輕震搖2h。然後將樹脂用DMF(3x3mL)和CH2Cl2(3x3mL)洗滌。偶聯效率通過Kaiser測試評價。對用Fmoc-Phe-OH和DOTA-(叔丁基酯)3-CO2H的兩個更多偶聯步驟重複上述順序。將最終化合物使用TFA/CH2Cl2(1/1)從樹脂上切割並在真空下濃縮以產生3。濃縮的產物通過使用C18SepPakVac2g柱純化。產物用70/30水/乙腈(在每個中的0.1%TFA)的溶液洗脫並凍幹。ESI-MS:974[M+H]+。向3(在1mLDMSO中的15mg,15.4μmol)的溶液添加1(15mg,26.18μmol)和TEA(30μL)並在環境溫度放置2h。在去除溶劑之後,化合物5通過HPLC(方法1)純化。1HNMR(DMSO-d6)δ:8.64(m,1H)、8.44(m,1H)、8.29-8.18(m,2H)、7.77-7.75(m,2H)、7.30-7.17(m,15H)、6.35-6.33(m,2H)、4.65-4.63(m,2H)、4.17-2.59(m,26)、2.40-1.11(m,30H)。13CNMR(DMSO-d6)δ:175.00、174.64、173.82、173.52、172.11、172.02、171.05、170.95、158.20、157.88、157.39、137.79、137.67、137.52、129.52、129.34、129.27、126.35、54.01、53.61、52.36、51.74、38.37、38.31、37.65、35.52、31.88、29.98、28.95、27.61、25.33、22.92、22.73。ESI-MS:1431[M+H]+,HRESI+-MS:對於C69H96N12O21,1431.7042[M+H]+的計算值,得出為:1431.7064。(21S,25S)-8,15,23-三氧代-1-((4-((1,4,7,10-四(羧甲基)-1,4,7,10-四氮雜環十二烷-2-基)甲基)苯基)氨基)-1-硫代-2,7,16,22,24-五氮雜二十七烷-21,25,27-三羧酸,6。化合物6如下文描述在三個步驟中進行製備。將商購可得的N-Boc-1,4二氨基丁烷(在0.5mlDMSO中的27mg,0.15mmol)與1,4,7,10-四氮雜環十二烷-1,4,7,10-四乙酸、2-[(4-異硫氰酸苯基)甲基](p-SCN-Bn-DOTA)(100mg,在1.5mLDMSO中的0.15mmol)和DIEA(132μl,0.75mmol)混合併在40℃攪拌4h。將溶劑蒸發,並將固體剩餘物使用水和乙腈(在各自中的0.1%TFA)通過反相C18快速層析(5.5g,AgilentSF10)純化,以在凍幹之後獲得Boc-保護的7。產率:~55%。ESI-MS740[M+H]+。然後將由該步驟得到的化合物用冰冷的TFA/CH2Cl2(1/1)溶液處理,並在環境溫度下放置攪拌2小時。將溶劑蒸發,並將剩餘物在真空下乾燥並通過反相快速層析(5.5g,AgilentSF10)純化,以中等產率產生7。1HNMR(DMSO-d6)δ:8.80-8.64(m,1H)、8.12-7.90(m,2H)、7.75-7.10(bm,4H)、4.65-4.63(m,1H)、4.17-2.59(m,27H)、2.40-1.11(m,6H)。ESI-MS:640[M+1]+。向7(在400μLDMSO中的11mg,17mol)的溶液添加1(在200μLDMSO中的10mg,17.4mol)和DIEA(27μL,170μmol),並在環境溫度放置2h。在蒸發溶劑之後,將剩餘物溶解於水中,並通過HPLC純化(方法2),以獲得6。Rt,22.5min。1HNMR(DMSO-d6)δ:8.88(m,1H)、8.44(m,1H)、8.21-7.98(m,2H)、7.77-7.75(m,2H)、6.35-6.33(m,2H)、4.65-4.63(m,2H)、4.17-2.59(m,29H)、2.40-1.11(m,30H)。HRESI-MS:對於C48H77N10O17S,1097.5183[M+H]+的計算值,得出為:1097.5212。[89Y]4。向4(在500μL0.5MNaOAc,pH6.8中的10mg,9.11μmol)的溶液添加50μL的YNO3(0.5M),並將混合物(pH6.1)在90℃孵育30min。添加EDTA(200μL,30mM,pH6.0)的溶液,並將反應混合物在40℃孵育10min,以絡合未反應的釔(III)。將所得化合物通過HPLC純化(方法2,Rt,21min),通過蒸發濃縮並凍幹。ESI-MS:1370[M+H]+。對於C60H87N11O20Y,1370.5187的計算值,得出為1370.5435。[89Y]5。HPLC純化通過方法2,Rt,26min,ESI-MS:1517[M+H]+。對於C69H96N12O21Y,[M+H]+1516.5793的計算值;得出為1516.5793。[89Y]6。HPLC,方法2,Rt,23min,HRESI+-MS。對於C48H77N10O17SY,1183.4007[M+H]+的計算值;得出為1183.4020。放射化學:[86Y]4-5和[86Y]6的放射性標記遵循與對於[86Y]6描述的相同的一般方法進行。[86Y]6。將新鮮製備的抗壞血酸(50μL,220μg)的溶液添加至86YNO3(在0.1M500μL硝酸中的111-148MBq(3-4mCi))的溶液以阻止放射分解。將在0.3MNaOAc中的約50-70μg的6(在N2下淨化2-3min)添加至該溶液,並通過加入μL3MNaOAc中和至pH~5.5-6,然後短暫渦旋混合物,該混合物隨後在95℃孵育20min。將反應混合物用1mL水稀釋。絡合作用通過將10-15μL溶液的等分試樣注入到HPLC來監測。如通過ITLC(GelmanITLC條,10mMEDTA)測量的,以~90-95%的放射化學產率獲得放射性標記的產物[86Y]6,其具有>98%的放射化學純度。對於作為混合物異構化合物的期望的產物獲得寬的放射性峰,Rt,~13.9-14.8min,並且對於游離配體的Rt為15.8min。比放射性>83.92GBq/μmol(n=5)。將酸性洗脫物用20μL1M碳酸鈉溶液中和,並在真空下將洗脫物的體積減少至乾燥。將固體剩餘物用鹽水稀釋至期望的放射性濃度,以用於生物分布和成像研究。有趣地,在中和和蒸發洗脫峰之後,在HPLC上再次注入示蹤物後,僅一個峰在約14.3min被分離。為了驗證[86Y]6的異構化,將化合物6用添加86Y的載體進行放射性標記,且混合物通過HPLC分析。僅一個峰在14.3min時被分離。對於[86Y]4-5,單個放射性標記的峰被分離。對於[86Y]4的Rt為14.0min,且對於未螯合的4的Rt為15.5min,而[86Y]5,Rt=16.9min,且對於未螯合的5Rt=19.5min。動物模型和測定:PSMA抑制活性使用基於螢光的測定(Banerjee,Pullambhatla,Byun,等,2011)確定。酶抑制常數(Ki值)使用Cheng-Prusoff轉化(Cheng和Prusoff,1973)產生。使用雄激素非依賴性PC-3人類前列腺癌異種移植物的亞系(Banerjee,Pullambhatla,Byun,等,2011)。那些亞系已被修飾以表達高(PC-3PIP)水平的PSMA或天然產生低(PC-3flu)水平的PSMA(WarrenHeston博士、ClevelandClinic,Cleveland,OH)。將表達PSMA的(PC-3PIP)和不表達PSMA的(PC-3flu)細胞系兩者在包含10%胎牛血清(FBS)(Invitrogen)和1%青黴素-鏈黴素(Pen-Strep)(Biofluids)的RPMI1640培養基(Invitrogen)中生長,如先前描述的(Banerjee,Pullambhatla,Byun,等,2011)。將6至8周齡雄性、非肥胖性糖尿病(NOD)/嚴重聯合免疫缺陷(SCID)小鼠(CharlesRiverLaboratories)分別在向頭側右脅腹和左脅腹處皮下(SC)植入PSMA+PC-3PIP和PSMA-PC-3flu細胞(在100μLMatrigel中的2x106個)。當異種移植物的直徑達到5mm至7mm時,將小鼠成像或用於生物分布測定。對於生物分布測定,攜帶PSMA+PC-3PIP和PSMA-PC-3flu異種移植物的NOD/SCID小鼠用0.55MBq(15μCi)86Y-4或86Y-6經由尾靜脈注射。在每種情況下,將四隻小鼠在注射後1h、2h、5h和24h時通過頸部脫位處死。將心臟、肺、肝、胃、胰腺、脾、脂肪、腎、肌肉、小腸和大腸、膀胱和PSMA+PC-3PIP和PSMA-PC-3腫瘤迅速取出。還收集了0.1-mL的血液樣品。將每個器官稱重,並且組織放射性用自動γ計數器(1282CompugammaCS,Pharmacia/LKBNuclearInc.)測量。每克組織注射劑量的百分比(%ID/g)使用連續稀釋的注射活性樣品計算。將所有活性測量值針對注射時間校正放射性衰變。動物成像:小動物PET和CT。對於成像研究,將攜帶PSMA+PC-3PIP和PSMA-PC-3flu腫瘤的NOD/SCID小鼠用3%異氟烷(v/v)麻醉,並保持在1.5%異氟烷(v/v)下。將小鼠(對於86Y-4或86Y-6的n=3及對於86Y-5的n=2)用在pH~7的100μL鹽水中製備的3.33-6.21MBq(90-168μCi)放射性示蹤物經由尾靜脈注射。對於結合特異性研究,在注射86Y-4之前30分鐘時,將小鼠皮下施用阻斷劑量的已知的PSMA抑制物N-[[[(S)-1-羧基-3-甲基丁基]氨基]羰基]-L-穀氨酸(ZJ43)(Olszewski等,2004)(50mg/kg),且另一小鼠用單獨的86Y-4注射。在不同的時間點,將麻醉的單獨的小鼠以俯臥位置放置在掃描儀臺架上並用醫用膠帶固定,同時將麻醉流速增加至0.8L/min。圖像使用FORE/2D-OSEM方法(兩次迭代,16個子集)重建,並包括對放射性衰變、掃描儀死時間和散射放射的校正。未進行部分體積校正(PVC)。在每次PET掃描之後,為解剖學共配準(anatomicco-registration)獲取CT掃描。為了促進PET和CT圖像共配準,採用了適合PET和CT掃描儀兩者的特殊動物床。當在掃描儀之間移動時以及在兩次掃描期間,動物處於麻醉下並固定化。重建的PET和CT圖像然後通過使用AMIDE軟體(來自sourceforge.net/amide)對準天然標誌(landmark)(諸如動物肢體和床輪廓)通過剛性變換來手動地共配準(co-registered)。數據使用AMIDE展示和分析。動態的全身PET和CT圖像分別在eXploreVISTA小動物PET(GEHealthcare,LittleChalfont,Buckinghamshire,UK)和X-SPECT小SPECT/CT系統(GammaMedicaIdeas,Northridge,CA)上獲取。86Y-6的東非狒狒(Papioanubis)(狒狒(baboon))PET成像。使用雄性東非狒狒(8歲,27.1kg)來研究86Y-6的生物分布。狒狒在圖像採集期間仰臥放置。對於衰減校正,在第一和最後PET成像之前立即獲取低劑量CT圖像。PET和CT圖像使用Hermes工作站(HermesMedicalSolutions,Greenville,SC)跨時間點共配準。將14個源器官輪廓藉助於疊加的PET/CT圖像在CT上描繪。從PET圖像中提取每個源器官的衰變校正的平均活性濃度(Bq/g)。對腎、腎皮質和前列腺的PET圖像繪製輪廓。對於前六個時間點(1h或更短),在PET圖像內定量的每器官衰變校正的總活性與施用的放射性幾乎一一對應,這證實了,所施用的放射性在PET圖像上被完全解釋,在這之後,由於排洩,總量小於施用量。在每個PET圖像中定量的放射性的總量用於獲得全身保留動力學。在作為快速推注式靜脈內施用80.7MBq(2.2mCi)的86Y-6之後,在5min、10min、15min、20min、35min、1h、2h、3.5h和23h時獲取9個靜態PET圖像。圖像在DiscoveryRxVCT掃描儀(GEHealthcare)上以2D模式獲取。輻射劑量學:對於每個時間點,活性濃度(以Bq/cm3計)在14個描繪的器官中的每一個中進行測量,並乘以器官體積以獲得每器官每時間點的總活性。然後將測量值進行衰減校正並除以狒狒器官質量(通過來自繪製的輪廓的CT密度和體積來確定)及注射的放射性,以獲得每個時間點和每個器官的每克初始放射性的分數(FIA/g)。然後,將狒狒FIA/g值使用以下等式(Schwartz等,2011;Woodard等,1975)轉化為人FIA(每器官):其中WB質量狒狒=27.1kg且WB質量人類=73.7kg.該方法假定,相對於全身中的總濃度,特定組織中的活性的濃度跨物種保持(即,對於狒狒和人類,器官濃度/總濃度相同)。將所得的人類FIA值作為對於每個器官的時間(9個數據點)的函數作圖,並擬合為雙指數表達式:其中A1、A2、λ1bio和λ2bio為擬合參數。A1和A2的總和給出了施用的放射性在每個器官中的反外推的、時間零分數,且λ1bio和λ2bio為生物學清除常數。如其名稱暗示的,通過積分等式(2)並引入物理衰減項(這取決於使用的同位素),獲得每個源器官的時間積分活性係數[TIAC,先前稱為停留時間(Bolch等,2009)等式:對90Y、177Lu和86Y計算TIAC,其中它們對應的物理衰變常數:=0.01083h-1(T1/2=64.0h);且分別排洩。放射吸收劑量通過使用OLINDA/EXM軟體(Stabin等,2005),根據MIRD吸收分數方法(Bolch等,2009)將時間積分活性轉換為吸收劑量來獲得。對於膀胱的TIAC使用在OLINDA/EXM中實現的MIRD膀胱模型來獲得。對該模型的輸入要求全身TIAC,其從擬合至全身保留動力學的等式獲得。將排洩間隔設置為2h。然後將TIAC輸入到OLINDA/EXM(Stabin等,2005),並且獲得14個器官的每單位放射性的所得的吸收劑量。使用OLINDA/EXM中的特定腎模型來獲得腎皮質劑量值。將來自腎臟外部器官的吸收劑量與從內腎模型計算的腎皮質劑量相加。特定的前列腺模型用於前列腺自身劑量,並且將膀胱的外部劑量與前列腺劑量相加,作為全身的替代物。對於唾液腺的每單位活性吸收劑量的自身劑量組分使用3D-RDMonteCarlo(EGSnrc)和具有描繪唾液腺的人類CT獲得。交叉劑量組分通過對相同大小的器官(胰腺)假設相同交叉劑量獲取。將測量的每器官每時間點活性濃度(以Bq/cm3計)值進行衰減校正並除以狒狒器官質量(通過來自繪製的輪廓的CT密度和體積來確定)及注射的放射性,以獲得每個時間點和每個器官的每克初始放射性的分數(FIA/g)。然後將狒狒FIA/g值使用相關等式轉化為人類FIA(每器官)(Olszewski等,2004;Schwartz等,2011)。然後將所得的人類FIA值作為時間的函數作圖,並擬合為雙指數表達式,並且計算每個源器官的對於時間積分活性係數(TIAC,先前稱為停留時間(Woodard等,1975))的值。放射吸收劑量通過使用OLINDA/EXM軟體(Bolch等,2009),根據MIRD吸收分數方法(Woodard等,1975)將時間積分活性轉換為吸收劑量來獲得。數據被表示為使用MicrosoftExcel(MicrosoftCorporation,2010)計算的平均值±標準差(SD)。使用Prism軟體(GraphPAD)以95%置信水平確定統計學顯著性,其中P≤0.05被認為是顯著的。結果化合物4和5使用如方案4和5中示出的組合的固相和溶液相肽合成策略製備。化合物1和4如先前報導的(Banerjee,Pullambhatla,Byun,等,2011)來製備。DOTA-綴合的配體5的合成根據方案5從Fmoc-Lys(Boc)-Wang樹脂開始,使用標準芴基甲氧基羰基(Fmoc)固相肽合成(SPPS)進行。將三個苯丙氨酸殘基與樹脂結合的賴氨酸偶聯,隨後DOTA綴合,在這之後,將化合物通過TFA/CH2Cl2的1/1混合物從樹脂上切割,以中等產率(~20%)產生3。然後將3的賴氨酸的游離ε-胺與1綴合(Davis等,2009)以產生5。化合物6通過在二異丙基乙胺的存在下在DMSO中使商購可得的DOTA-苄基-異氰酸酯和N-Boc-1,4-二氨基丁烷在40℃反應4h來合成,然後去除Boc基團,以在通過HPLC純化之後以中等產率產生7。然後將化合物7與1綴合,以良好產率產生6。如方案4-5中示出的,穩定的釔(89Y)絡合物通過在95℃將綴合物4-6與YNO3的水性溶液孵育來製備。值得提及的是,86/89Y(III)-標記的化合物4-5包含與金屬配位的三個羧酸,使得其為總體中性化合物,而[86/89Y]6具有四個配位的羧酸,形成總體帶負電荷的化合物。放射性示蹤物[86Y]4-6通過在pH5-6的沸水中在10-6M的配體濃度下與[86Y]NO3反應30min後,使用相同的一般程序製備。a.(i)20%哌啶/DMF;(ii)Fmoc-Phe-OH、HOBT、HBTU,步驟i-ii對於X=2重複2次,且對於x=3重複3次,(iii)20%哌啶/DMF;(iv)DOTA-三(叔丁基酯)-CO2H,HOBT,HBTU,DIEA;(v)TFA/TES/H2O(98/0.5/1.5);b.DMSO/TEA,rt;c.Y(NO3)3/NaOAc,pH5.5,90℃,20min;d.86Y(NO3)3/抗壞血酸/NaOAc,pH5.5,90℃,20min方案4.4-5和[86/89Y]4-5的合成。方案5.6和[86/89Y]6的合成。86Y-標記的PSMA靶向化合物86Y-4、86Y-5和86Y-6的化學結構示於圖14中。靶化合物的放射性標記以高產率(~90-97%)和放射化學純度(>98%)與高比放射性(>83.92GBq/μmol(2.27Ci/μmol))進行。所有化合物展示出高結合親和力,其中Ki值範圍為從0.10nM至4.69nM(表2)。表2.PSMA抑制活性小動物PET成像:對86Y-4、86Y-5和86Y-6獲得全身PET/CT圖像(圖18A、圖18B、圖18C、圖19A、圖20A、圖20B和圖20C)。在注射後2h時,所有三种放射性示蹤物使得能夠可視化PSMA+PC-3PIP腫瘤和腎(已知表達PSMA的器官)(圖18A、圖18B和圖18C)。放射性示蹤物的腎攝取部分地是由於這些試劑的排洩途徑以及由於從小鼠近端腎小管中PSMA的表達的特異性攝取(Stabin等,2005)。試劑86Y-5在胃腸道中顯示出非特異性累積,推測是由於連接基部分上的三個Phe殘基的疏水性增加。考慮到這類低分子量化合物的短生物半衰期,86Y-4的PET-CT圖像在注射後1h、4h和18h時獲得。多達至4h觀察到PSMA+PC-3PIP腫瘤和腎和膀胱中放射性示蹤物的存在(圖19A)。儘管PSMA+PC-3PIP腫瘤保留一些活性,到18h時,膀胱和腎中的放射性顯著清除。作為體內結合特異性的進一步測試,86Y-4的阻斷研究通過用50mg/kg強效的、選擇性PSMA抑制物ZJ43(Silver等,1997)預處理動物來進行。圖19B顯示了,ZJ43不僅在腫瘤內而且在腎皮質(另一個表達PSMA的組織)內能夠阻斷86Y-4的結合(Stabin等,2005)。圖20A、圖20B和圖20C展示在注射後0.5h、2h和12h時對於86Y-6的PET-CT成像。顯著地,86Y-6表現出放射性從正常組織的更快清除,並且到注射後12h時,放射性被大量地從腎清除,產生清楚的腫瘤對背景對比。早在15min時實現了PSMA+PC-3PIP腫瘤的清楚描繪。顯著地,86Y-6不包含86Y-4和86Y-5的另外的苯丙氨酸部分,並且使用對異硫氰酸根苄基1,4,7,10-四氮雜環十二烷-1,4,7,10-四乙酸(DOTA)螯合物,其添加額外的羧酸鹽以牢固地保持金屬並降低親脂性。小鼠中的生物分布:基於成像的結果,化合物86Y-4和86Y-6在標準生物分布測定(Banerjee、Pullambhatla、Byun,等,2011)中被進一步評價。表3和4示出了在注射後1h、2h、5h和24h時選擇的器官中的%ID/g攝取值。兩种放射性示蹤物在PSMA+PC-3PIP腫瘤異種移植物中顯示PSMA依賴性結合,其中86Y-4在早在注射後1h時顯示高的腫瘤攝取(29.3±8.7%ID/g),具有相對慢的清除率,在注射後5h為15.7±1.7%ID/g且在24h時為5.9±0.8%ID/g。PSMA+PC-3PIP腫瘤對PSMA-PC-3flu腫瘤攝取比率範圍為從1h時的89至24h時的高的229。血液和正常組織諸如心臟、肝、胃和胰腺在24h之後未顯示顯著的攝取(~1%ID/g)且降低至低於0.02%ID/g。PSMA+PC-3PIP腫瘤與肌肉的比率也很高,在24h時達到最大值為1,046。發現腎攝取果然是高的並且在1h時達到峰值244.9±8.8%ID/g且到24h時降低至1.5±0.7%ID/g。表4示出了對於86Y-6的器官%ID/g攝取值。化合物86Y-6在注射後1h內在PSMA+PC-3PIP腫瘤內迅速累積,其中攝取值為26.6±1.9%ID/g。放射性示蹤物濃度在PSMA+PC-3PIP腫瘤內持續增加,在注射後5h時表現32.2±8.0%ID/g的最高攝取。腫瘤攝取保持高的直至注射後24h。正常器官諸如血液、心臟、肝、脾、胃和胰腺在1h時表現出低攝取,其到5h時降低至低於0.4%ID/g。對於86Y-6的腎攝取在1h和2h時分別為86.5±13.6%ID/g和54.0±9.2%ID/g,比對於86Y-4的低得多。表3.86Y-4在小鼠中的生物分布(%ID/g)表4.86Y-6在小鼠中的生物分布(%ID/g)1H2H5H24H血液0.6±0.00.2±0.00.1±0.00.0±0.0心臟0.3±0.00.1±0.00.0±0.00.0±0.0肺1.1±0.20.5±0.10.2±0.00.1±0.0肝0.3±0.00.2±0.00.1±0.00.1±0.0胃0.3±0.10.14±0.010.11±0.010.05±0.09胰腺0.3±0.10.23±0.20.08±0.040.01±0.01脾3.0±0.71.31±0.70.36±0.120.11±0.05脂肪0.6±0.51.87±3.440.12±0.170.01±0.01腎87.0±14.054.0±9.015.6±4.14.8±0.8肌肉0.8±1.20.25±0.20.0±0.00.0±0.0小腸0.3±0.10.1±0.00.07±0.020.02±0.02大腸0.4±0.30.2±0.10.1±00.0±0.0膀胱6.0±3.95.5±3.70.8±0.10.4±0.3PC-3PIP26.6±1.929.2±2.332.2±8.015.8±6.4PC-3flu0.4±0.10.2±0.00.2±0.10.1±0.1PIP:flu66152183130PIP:血液44145378620PIP:肌肉331159213,01086Y-6的狒狒PeT成像和藥代動力學:圖21A和圖21B描繪了PET研究,其中在肝、唾液腺、腎和膀胱中觀察到放射性示蹤物。對於全腎、腎皮質和前列腺,在每個PET圖像上繪製輪廓用於定量。所有器官顯示兩相(快速和緩慢)生物學清除。腎在注射後約25min時具有最高攝取(8%ID/g)。在腎中觀察到的放射性的68%以約1h(0.84h)的生物半衰期被清除,且剩餘的放射性以16.6h的生物半衰期被清除。腎皮質中的大部分(66%)放射性以1.1h的生物半衰期被清除,且剩餘的放射性以約19h的生物半衰期被清除。儘管與用68Ga-標記的PSMA靶向的試劑和124/131I-MIP.1095(zechmann等,2014)成像的患者的PET掃描相比為中度的,在肝和唾液腺中觀察到顯著的攝取和保留。表5給出了所有器官的生物清除動力學的小結。在劑量計算中使用的TIAC列於表6中。表5.[86Y]6擬合的藥代動力學參數表6.時間積分活性係數(停留時間)器官吸收劑量:表7提供了對於86Y、90Y/177Lu的器官吸收劑量的詳細列表,以mGy/MBq的單位表示。對於所有同位素,腎皮質接收到最高的每單位活性吸收劑量。因此,可能的是,在患者特異性吸收劑量治療計劃(Baechler等,2012;Hobbs等,2009)的背景下,腎皮質將是用於治療性放射性金屬的劑量限制器官,隨後是膀胱。對於診斷同位素86Y,0.099mSv/MBq的有效劑量也在OLINDA/EXM中進行計算。表7.基於狒狒PET成像數據的參考成年男性的器官吸收劑量討論三種86Y-標記的PSMA-靶向的試劑已被合成並評價,以便進行非人類靈長類劑量學。這些化合物包含與已公開的其他化合物相似的與靶向脲附連的DOTA-或DOTA單醯胺螯合的放射性金屬(Banerjee等,2010;Banerjee、Pullambhatla、Byun,等,2011)。由於DOTA及其衍生物兩者都可用於PET(86Y)或放射性藥物療法(90Y),它們一直是焦點。已經證明,藥代動力學取決於所用的放射性金屬螯合物,包括特別設計用於結合PSMA的化合物的那些。不希望受任何一種特定理論所束縛,認為這主要歸因於放射性配體的總電荷和金屬螯合絡合物的穩定性。特別地,在68Ga-標記的PSMA-結合DOTA綴合的試劑的以前的報導中,68Ga-4顯示從正常組織,包括腎的最快清除(Banerjee等,2010)。然而,在目前的研究中,觀察到86Y-4出乎意料地表現出較高的腎吸收。對86Y-6的評價顯示了放射療法所需的期望的較低的腎攝取和較高的腫瘤保留,並且被隨後選擇用於在狒狒中定量PET成像,以用於劑量學測量。結合特異性研究(圖19B)指示,在1小時時,幾乎所有的86Y-4的腎結合是特異性的而不是由於排洩。證據顯示,與腫瘤相比,腎實質中更有組織且快速的血流可能解釋許多這些試劑的較長久的腫瘤而不是腎保留。儘管PSMA結合親和力為可能確定腫瘤對比腎攝取的一個因素,其他因素,諸如親脂性、電荷、血漿蛋白結合和分子量也可能起重要作用。對於90Y為1.19mGy/MBq和對於177Lu為0.245mGy/MBq的估計腎皮質劑量與在涉及肽受體放射療法的報導中計算的對於90Y為1.97mGy/MBq和對於177Lu為0.45mGy/MBq的值(其中腎皮質是劑量限制器官)相比有利(Baechler等,2012)。由於已知DOTA和許多DOTA衍生物形成動力學和熱力學穩定的絡合物,常用的和臨床實施的螯合劑DOTA用於所有三种放射性配體。在許多情況下,已顯示對應的Y(III)-絡合物在體內是穩定的,這是螯合物的期望性狀。顯著地,還報導了,DOTA與包括鑭系元素(例如,177Lu(III))和錒系元素(例如,225Ac(III))的一批三價金屬離子(與86Y(III)化學上不同)形成穩定的絡合物。此外,結合PSMA的基於脲的試劑在用於DOTA的放射性標記條件下是穩定的。最近,PSMA-靶向的單克隆抗體J591的90Y或177Lu-標記的版本在階段I和II臨床試驗中顯示有希望的結果(Bander等,2005;Tagawa、Akhtar,等,2013;Tagawa、Milowsky,等,2013)。在那些情況下,111In-標記的抗體用於劑量學計算(Vallabhajosula等,2005)。儘管那些放射性標記的單克隆抗體具有腫瘤檢測和療法的潛力,其適度的腫瘤靶向和對紅色骨髓的相對高的吸收劑量減輕了對常規臨床使用。作為一種可替代方法,使用131I-標記的PSMA-靶向的、基於脲的小分子的早期臨床結果顯示高劑量遞送至惡性病灶(Zechmann等,2014)。在那些公布的研究中,唾液腺顯示出最高的吸收劑量(4.62mGy/MBq),隨後是肝(1.47mGy/MBq)和腎(1.45mGy/MBq)二者(Zechmann等,2014)。如也由相對高(0.91mGy/MBq)的甲狀腺吸收劑量證明的,唾液腺吸收劑量的顯著貢獻為游離碘攝取是可能的,這在本研究中不發生。通常,對於86Y-6,從正常器官的清除率比公布的結果(Zechmann等,2014)更快,腎例外。總之,生物分布和劑量學結果表明,86Y-6為用於表達PSMA的腫瘤的定量PET成像的有希望的候選者,並且可提供用於計劃和監測PSMA-靶向的基於90Y-、177Lu-的放射性藥物療法的適合的成像替代物。實施例4用於基於PSMA的靶向性放射性核素療法的177Lu-SR-VI-71、203Pb-SR-VI-71和203Pb-SR-IX-11圖24,使用0.01-10μCi的177Lu-SRVI71的細胞攝取研究顯示,在PSMA+PIP腫瘤中的高攝取和在PSMA-flu腫瘤中的可忽略不計的攝取。另外,內化研究顯示,總細胞相關放射性的~44%被內化。此外,在10μM的N-[[[(1S)-1-羧基-3-甲基丁基]氨基]羰基]-L-穀氨酸(ZJ43)(PSMA的一種特定抑制物)的共孵育後觀察到PSMA+細胞中~90%阻斷,進一步確認了試劑的優異特異性(圖23)。體內評價在標準PSMA+PIP和PSMA-flu小鼠異種移植物中並通過用VECT或具有額外超高靈敏度小鼠準直器的儀器進行SPECT成像來進行,如圖25A、圖25B和圖25C公開的。在所有時間點在PSMA+PIP腫瘤中發現放射性的最高累積。其他可見器官為腎和膀胱。在2h(59.1±12.8%ID/g)和在24h(40.6±5.8%ID/g)時的生物分布研究(n=4)實際上顯示了PSMA+腫瘤中以高特異性的高攝取和保留(在2h時在~180的PIP:flu)。在2h時,初始腎攝取高達89.3±28.9%,隨後在24h內快速清除(6.29±3.4%ID/g)。表8.177Lu-SR-VI-71的組織生物分布表9.203Pb-SR-VI-71的組織生物分布表10.在2h時203Pb-SR-IX-11(n=2)和203Pb-SR-VI-71(n=4)的組織生物分布203Pb-SR-IX-11203Pb-SR-VI-71血液0.07±0.010.31±0.04心臟0.07±0.010.14±0.06肺0.24±0.030.64±0.14肝0.21±0.011.01±0.21胃0.12±0.000.15±0.03胰腺0.06±0.000.29±0.05脾0.25±0.11.59±0.58脂肪1.54±1.640.31±0.22腎3.61±0.8139.35±7.28肌肉0.78±0.830.22±0.07小腸0.25±0.150.23±0.04唾液腺0.35±0.100.93±0.07膀胱8.64±0.7710.40±3.21PC-3PIP21.64±1.2338.14±6.30PC-3flu0.19±0.040.29±0.14PIP:flu114130這些結果在製備具有用於放射性核素療法的期望的藥代動力學的此類低分子量治療劑的可行性方面是相當有希望的。數據進一步支持,這類LMW劑可在與PSMA結合後有效內化。實施例5用於基於PSMA的靶向的放射性核素療法的ZCP-01和相關的試劑的合成和使用概述用於表達PSMA的腫瘤的成像和可能的放射療法的經由多種連接基基團與螯合的放射性金屬綴合的PSMA結合脲的製備和使用已在幾種專利和出版物(Banerjee,等,2008;Banerjee,等,2010;Banerjee,等,2011;Banerjee,等,Oncotarget2011;Banerjee,等,2013;Banerjee,等,2014)以及本專利申請中被描述。最近已開發了,基於對應於氨基甲酸酯支架的新穎賴氨酸-氨基甲酸酯支架氧代戊二酸(OPA)和對應於「反向」氨基甲酸酯支架的氨基-戊二酸(NPA)構建的PSMA抑制物,包括F-18標記的類似物,如圖29上公開的。F-18標記的NPA和OPA化合物在PSMA陽性腫瘤小鼠異種移植物中顯示選擇性攝取。ZCP-01,用於PSMA陽性腫瘤和組織的成像和放射療法的用於絡合放射性金屬的DOTA-PEG-連接的賴氨酸OPA氨基甲酸酯已被合成作為實例。先前在專利申請第WO2009/002529A2號和第WO2010/108125A2號中公開的用於與脲一起使用的寬範圍的金屬螯合配體和連接基可附連至OPA和NPA支架,以提供用於前列腺癌的成像和/或放射療法的新型放射性標記的試劑。材料和方法(18S,22S)-2,12,20-三氧代-1-(4,7,10-三(羧甲基)-1,4,7,10-四氮雜環十二烷-1-基)-6,9,21-三氧雜-3,13,19-三氮雜二十四烷-18,22,24-三羧酸,ZCP-01。參考方案7,向化合物4(在200μLDMSO中的8.5mg,0.015mmol)的溶液添加二異丙基乙胺(27μL,0.255mmol),並隨後緩慢添加DOTA-NHS(在200μLDMSO中的15.2mg,0.023mmol),並將所得溶液在室溫下保持攪拌2h。然後將溶液用水稀釋並通過HPLC純化。HPLC方法:PhenomenexC18Luna,10mm*250mm,流速:8ml/min,λ:200nm,220nm,溶劑H2O和CH3CN(在每個中的0.1%TFA)。梯度法;0-20min,100/0H2O/CH3CN至80/20H2O/CH3CN;20-30min80/20H2O/CH3CN至0/100H2O/CH3CN;31min100/0H2O/CH3CN。HPLC保留時間(tr)=16min.ESI-MS:954(M+H)。產率:在HPLC純化之後為9.4mg(~65.7%)。113/115In-ZPC-01的製備向ZPC-01(在500μL0.5MNaOAc,pH6.8中的5mg,5.24μmol)的溶液添加50μL的InNO3(0.5M),並將混合物(pH6)在90℃孵育30min。添加EDTA(200μL,30mM,pH6.0)的溶液,並將反應混合物在40℃孵育10min,以絡合未反應的銦(III)。將所得化合物通過HPLC純化(與ZPC-01相同),通過蒸發濃縮並凍幹。ESI-MS:1066[M+H]+。對於C39H64InN7O20,計算為1065.79。111In-ZPC-01的製備。將1.0μl在0.1NHCl中的111InCl3(1mCi)添加至20μl在0.2MNaOAc中的1mMOurea-PEG-DOTA。混合物的pH為~4.0。然後,20μl0.2MNaOAc以將pH調節為~6。將混合物在50℃保持1小時,並使用包含流動相90%水(包含0.1%TFA)和10%CH3CN(0.1%TFA)的等度方法,通過放射性-HPLC純化;流速:1.0mL/min;λ:200nm,和C18柱(25×4.6mm),Varianmicrosob-MV100-5。放射性標記的[111In]ZPC-01在14.9min時洗脫,而未標記的螯合劑在32min時洗脫。方案7a.DCC,N-羥基琥珀醯亞胺,CH2Cl2;b.二異丙基乙胺,DMSO;TFA/水;d.DO3A-NHS(從Macrocylics商購可得)。結果ZCP-01和[In]-ZCP-01展示高結合親和力,其中Ki值範圍分別為從17.82nM至58.21nM和0.29μM至0.92μM(表8)。表11.對於ZCP-01和[In]-ZCP-01的PSMA抑制數據在靜脈注射[111In]-ZCP-01之後,對攜帶分別在右脅腹和左脅腹皮下植入的PSMA+PC3PIP和PSMA-PC3flu腫瘤異種移植物的小鼠進行[In]-ZCP-01的體內SPECT成像,如圖34A、圖34B和圖34C上示出的。然而,[111In]-ZCP-01使得能夠在注射後2h和4h時可視化PSMA+PC3PIP腫瘤和腎(已知的表達PSMA的器官),而flu腫瘤接受非特異性攝取。到注射後24h時,放射性被大量地從腫瘤和腎清除。這些結果在製備具有用於腫瘤的成像和放射療法的期望的藥代動力學的此類低分子量治療劑的可行性方面是相當有希望的。參考文獻在說明書中提到的所有出版物、專利申請、專利和其他參考文獻表示本發明公開的主題所屬領域的技術人員的水平。所有出版物、專利申請、專利和其他參考文獻在此通過引用併入,其程度如同每個單獨出版物、專利申請、專利和其他參考文獻被具體和單獨指明通過引用併入的相同程度。將理解,儘管一些專利申請、專利和其他參考文獻在本文中被提及,此類參考文獻並不構成任何這些文件形成本領域公知常識的部分的承認。2008/12/31公布的用於Labeledinhibitorsofprostatespecificmembraneantigen(PSMA),biologicalevaluation,anduseasimagingagents的屬於Pomper,M.G.,Ray,S.,Mease,R.C.,Foss,C的國際PCT專利申請公布第PCT/US2008/007947號(WO2009/002529A2).2010/09/23公布的用於PSMA-targetingcompoundsandusesthereof的屬於Pomper,M.G.,Mease,R.C.;Ray,S.,Chen,Y.的國際PCT專利申請公布第PCT/US2010/028020號(WO2010/108125A2).2009/06/04公布的用於Prostatespecificmembraneantigen(PSMA)targetednanoparticlesfortherapyofprostatecancer的屬於Chandran,S.S.,Ray,S.,Denmeade,S.R.,Pomper,M.G.,Mease,R.C.的國際PCT專利申請公布第PCT/US2008/013158號(WO2009/070302A1).Aime,S.,etal.CurrPharmBiotechnol2004,5,509-518.Artemov,D.Molecularmagneticresonanceimagingwithtargetedcontrastagents.J.Cell.Biochem.Oct152003;90(3):518-524.Artemov,D.,MoriN.,Okollie,B.,Bhujwalla,Z.M.MagnResonMed2003,49,403-408.Artemov,D.,MoriN.,Ravi,R.,Bhujwalla,Z.M.CancerRes2003,63,2723-2727.Babich,J.W.,Zimmerman,C.,Joyal,J.,Lu,G.Radiolabeledprostatespecificmembraneantigeninhibitors.US2013/0034494A1.Baechler,S.,etal.Three-dimensionalradiobiologicaldosimetryofkidneysfortreatmentplanninginpeptidereceptorradionuclidetherapy.Med.Phys.2012;39:6118-6128.Bander,N.H.,etal.PhaseItrialof177lutetium-labeledJ591,amonoclonalantibodytoprostate-specificmembraneantigen,inpatientswithandrogen-independentprostatecancer.J.Clin.Oncol.2005;23:4591-4601.Banerjee,S.R.,etal.Synthesisandevaluationoftechnetium-99m-andrhenium-labeledinhibitotsoftheprostate-specificmembraneantigen(PSMA).J.Med.Chem.2008;51:4504-4517.Banerjee,S.R.,Pullambhatla,M.,Byun,Y.,etal.68Ga-labeledinhibitotsofprostate-specificmembraneantigen(PSMA)forimagingprostatecancer.J.Med.Chem.2010;53:5333-5341.Banerjee,S.R.,Pullambhatla,M.,Shallal,H.,etal.SequentialSPECTandOpticalImagingofExperimentalModelsofProstateCancerwithaDualModalityInhibitoroftheProstate-SpecificMembraneAntigen.Angew.Chem.Int.Ed.Engl.Sep192011;50(39):9167-9170.Banerjee,S.R.,etal.Amodularstrategytopreparemultivalentinhibitorsofprostate-specificmembraneantigen(PSMA).Oncotarget.2011;2:1244-1253.Banergee,S.R.,Pullambhatla,M.,Foss,C.A.,Falk,A.,Byun,Y.,Nimmagadda,S.,Mease,R.C.,Pomper,M.G.Effectofchelatorsonthepharmacokineticsof99mTc-labeledimagingagentsfortheprostate-specificmembraneantigen(PSMA).J.Med.Chem.2013;56:6108-6121.Banergee,S.R.,Pullambhatla,M.,Foss,C.A.,Nimmagadda,S.,Ferdani,R.,Anderson,C.J.,Mease,R.C.,Pomper,M.G.64Cu-Labeledinhibitorsofprostate-specificmembraneantigenforPETimagingofprostatecancer.J.Med.Chem.2014;57:2657-2669.Banerjee,S.R.,Foss,C.A.,Pullambhatla,M.,Wang,Y.,Srinivasan,S.R.F.KE.Hobbs,M.Baidoo,R.C.Brechbiel,Mease,G.Sgouros,M.G.Pomper,Preclinicalevaluationof86Y-labeledinhibitorsofprostatespecificmembraneantigenfordosimetryestimates.JNuclMed2015.Baur,B.,etal.Synthesis,radiolabellingandinvitrocharacterizationoftheGallium-68-,Yttrium-90-andLutetium-177-labelledPSMAligand,CHX-A″-DTPA-DUPA-Pep.Pharmaceuticals.2014;7:517-529.BehnamAzad,B.,etal.Nanoscale2015,7,4432-4442.Bodei,L.,etal.Receptorradionuclidetherapywith90Y-[DOTA]0-Tyr3-octreotide(90Y-DOTATOC)inneuroendocrinetumours.Eur.J.Nucl.Med.Mol.Imaging.2004;31:1038-1046.Bolch,W.E.,etal.,MIRDpamphletNo.21:ageneralizedschemaforradiopharmaceuticaldosimetry--standardizationofnomenclature.J.Nucl.Med.2009;50:477-484.Boros,E.,etal.JAmChemSoc2012,134,19858-19868.Caravan,P.,etal.InorgChem2007,46,6632-6639.Caravan,P.,etal.ContrastMediaMolImaging2009,4,89-100.Chandran,S.S.,etal.,Characterizationofatargetednanoparticlefunctionalizedwithaurea-basedinhibitorofprostate-specificmembraneantigen(PSMA).CancerBiol.Therapy.2008;7:974-982.Chappell,L.L.etal.Synthesis,characterization,andevaluationofanovelbifunctionalchelatingagentfortheleadisotopes203Pband212Pb,NuclMedBiol,27(2000)93-100.Chen,Z.,Penet,etal.PSMA-targetedtheranosticnanoplexforprostatecancertherapy.ACSNano.2012;6:7752-7762.Cheng,Y.andPrusoff,W.H.Relationshipbetweentheinhibitionconstant(K1)andtheconcentrationofinhibitorwhichcauses50percentinhibition(150)ofanenzymaticreaction.Biochem.Pharmacol.1973;22:3099-3108.Cho,S.Y.etal.Biodistribution,tumordetection,andradiationdosimetryof18F-DCFBC,alow-molecular-weightinhibitorofprostate-specificmembraneantigen,inpatientswithmetastaticprostatecancer.J.Nucl.Med.2012;53:1883-1891.Davis,S.L.,etal.Bacterialthymidinekinaseasanon-invasiveimagingreporterforMycobacteriumtuberculosisinliveanimals.PloSone2009;4:e6297.DeLeon-Rodriguez,L.M.,etal.MRIdetectionofVEGFR2invivousingalowmolecularweightpeptoid-(Gd)8-dendronfortargeting.J.Am.Chem.Soc.Sep222010;132(37):12829-12831.Evans,M.J.,etal.ProcNatlAcadSciUSA2011,108,9578-9582.Frenzel,T.,etal.InvestRadiol2008,43,817-828.Geninatti-Crich,S.ContrastMediaMolImaging2011,6,421-425.Ghosh,A.andHeston,W.D.Tumortargetprostatespecificmembraneantigen(PSMA)anditsregulationinprostatecancer.J.Cell.Biochem.2004;91:528-539.Haffner,M.C.,etal.HumPathol2009,40,1754-1761Haffner,M.C.,etal.ModPathol2012,25,1079-1085.Hanaoka,K.,etal.MagnResonImaging2008,26,608-617.Helisch,A.,etal.Pre-therapeuticdosimetryandbiodistributionof86Y-DOTAPhel-Tyr3-octreotideversus111In-pentetreotideinpatientswithadvancedneuroendocrinetumours.Eur.J.Nucl.Med.Mol.Imaging.2004;31:1386-1392.Hillier,S.M.,etal.JNuclMed2011,52,1087-1093.Hobbs,R.F.,etal.124IPET-based3D-RDdosimetryforapediatricthyroidcancerpatient:real-timetreatmentplanningandmethodologiccomparison.J.Nucl.Med.2009;50:1844-1847.Huang,C.H.andTsourkas,A.CurrTopMedChem2013,13,411-421.Kaiser,E.,etal.Colortestfordetectionoffreeterminalaminogroupsinthesolid-phasesynthesisofpeptides.AnalBiochem1970;34:595-598.Kam,B.L.,etal.Lutetium-labelledpeptidesfortherapyofneuroendocrinetumours.Eur.J.Nucl.Med.Mol.Imaging.2012;39Suppl1:S103-112.Konda,etal.Magma2001,12,104-113.Kulkarni,H.M.W.,etal.FirstclinicalresultswithLu-177PSMA-TUM1forthetreatmentofcastrate-resistantmetastaticprostatecancer.J.NUCL.Med.MeetingAbstractVol55;2014.Lanza,M.,etal.JNuclCardioL2004,11,733-743.Low,P.S.,Chelvam,V.,Kim,Y.PSMAbindinglinkerconjugatesandmethodsforusingWO2011/106639,WO2010/045598A2,WO2009/026177A1.Ma,D.,etal.RadioimmunotherapyformodelBcellmalignanciesusing90Y-labeledanti-CD19andanti-CD20monoclonalantibodies.Leukemia.2002;16:60-66.Major,J.L.andMeade,T.J.AccChemRes2009,42,893-903.Mastarone,D.J.,etal.Amodularsystemforthesynthesisofmultiplexedmagneticresonanceprobes.JAmChemSoc.Apr132011;133(14):5329-5337.McDevitt,M.R.Radioimmunotherapywithalpha-emittingnuclides,EurJNuclMed,25(1998)1341-1351.Mease,R.C.,etal.PETimaginginprostatecaneer:focusonprostate-specificmembraneantigen.Curr.Top.Med.Chem.2013;13:951-962.Milowsky,M.I.,etal.Vasculartargetedtherapywithanti-prostatespecificmembraneantigenmonoclonalantibodyJ591inadvancedsolidtumors.J.Clin.Oncol.2007;25:540-547.Nayak,T.K.andBrechbiel,M.W.86YbasedPETradiopharmaceuticals:radiochemistryandbiologicalapplications.Med.Chem.2011;7:380-388.Olson,W.C.,etal.Clinicaltrialsofcaneertherapiestargetingprostatespecificmembraneantigen.Rev.RecentClin.Trials.2007;2:182-190.Olszewski,R.T.,etal.NAAGpeptidaseinhibitionreduceslocomotoractivityandsomestereotypesinthePCPmodelofschizophreniaviagroupIImGluR.J.Neurochem.2004;89:876-885.Palm,S.,etal.Pharmacokineticsandbiodistributionof(86)Ytrastuzumabfor(90)Ydosimetryinanovariancarcinomamodel:correlativeMicroPETandMRI.J.NuclMed.2003;44:1148-1155.Pomper,M.G.,Ray,S.,Mease,R.C.,Foss,C.Labeledinhibitorsofprostatespecificmembraneantigen(PSMA),biologicalevaluation,anduseasimagingagents.WO2009/002529A2.Rooney,W.D.,etal.MagnResonMed2007,57,308-318.Schulke,N.,etal.Thehomodimerofprostate-specificmembraneantigenisafunctionaltargetforcancertherapy.Proc.Natl.Acad..Sci.USA.2003;100:12590-12595.Schwartz,J.,etal.Renaluptakeofbismuth-213anditscontributiontokidneyradiationdosefollowingadministrationofactinium-225-labeledantibody.Phys.Med.Biol.2011;56:721-733.Silver,D.A.,etal.Prostate-specificmembraneantigenexpressioninnormalandmaIignanthumantissues.Clin.CancerRes.1997;3:81-85.Song,Y.,etal.JAmChemSoc2008,130,6662-6663.Stabin,M.G.,etal.OLINDA/EXM:thesecond-generationpersonalcomputersoftwareforinternaldoseassessmentinnuclearmedicine.J.Nucl.Med.2005;46:1023-1027.Tagawa,S.T.,etal.Bonemarrowrecoveryandsubsequentchemotherapyfollowingradiolabeledanti-prostate-specificmembraneantigenmonoclonalantibodyj591inmenwithmetastaticcastration-resistantprostatecancer.Front.Oncol.2013;3:1-6.Tagawa,S.T.,etal.PhaseIIstudyofLutetium-177-labeledanti-prostatespecificmembraneantigenmonoclonalantibodyJ591formetastaticcastration-resistantprostatecancer.Clin.CancerRes.2013;19:5182-5191.Tse,B.W.,etal.Nanomedicine2015,10,375-386.Vallabhajosula,S.,etal.Pharmacokineticsandbiodistributionof111In-and177Lu-labeledJ591antibodyspecificforprostate-specificmembraneantigen:predictionof90YJ591radiationdosimetrybasedon111Inor177Lu?J.Nucl.Med.2005;46:634-641.Witzig,T.E.,etal.Safetyofyttrium-90ibritumomabtiuxetanradioimmunotherapyforrelapsedlow-grade,follicular,ortransformednon-Hodgkin′slymphoma.J.Clin.Oncol.2003;21:1263-1270.Woodard,H.Q.,etal.Letter:Expressionoftissueisotopedistribution.J.Nucl.Med.1975;16:958-959.Wu,X.etal.BioconjugChem2012;23,1548-1556.Yong,K.andBrechbiel,M.W.Towardstranslationof212Pbasaclinicaltherapeutic;gettingtheleadin!,DaltonTrans,40(2011)6068-6076.Yong,K.J.etal.(212)Pb-radioimmunotherapyinducesG(2)cell-cyclearrestanddelaysDNAdamagerepairintumorxenograftsinamodelfordisseminatedintraperitonealdisease,Mol.CancerTher.,11(2012)639-648.Yong,K.J.etal.212Pb-radioimmunotherapypotentiatespaclitaxel-inducedcellkillingefficacybyperturbingthemitoticspindlecheckpoint,Br.J.Cancer,108(2013)2013-2020.Yoo,J.,etal.Preparationofhighspecificactivity(86)Yusingasmallbiomedicalcyclotron.Nucl.Med.Biol.2005;32:891-897.Zechmann,C.M.,etal.Radiationdosimetryandfirsttherapyresultswitha(124)I/(131)I-labeledsmallmolecule(MIP-1095)targetingPSMAforprostatecancertherapy.Eur.J.Nucl.Med.Mol.Imaging.2014;41:1280-292.儘管為了清楚理解的目的通過說明和實施例相當詳細地描述了前述主題,本領域技術人員將理解,可在所附權利要求的範圍內實踐某些改變和修改。當前第1頁1 2 3