聚醯亞胺薄膜、使用該聚醯亞胺薄膜的有機電致發光元件以及有機電致發光顯示器的製作方法
2024-01-21 11:19:15 2
本發明涉及聚醯亞胺薄膜、使用該聚醯亞胺薄膜的有機電致發光元件以及有機電致發光顯示器。
背景技術:
近年來,在使用有機電致發光元件的顯示器以及液晶顯示器等的顯示器設備領域等中,需求具有如玻璃般的高透光性且充分高的耐熱性,並且又輕又柔軟的材料。作為該用於替代玻璃等的材料,現有技術著眼於由具有高耐熱性、又輕又柔軟的聚醯亞胺所形成的薄膜。
該聚醯亞胺公知如芳香族聚醯亞胺(例如dupont公司制的商品名「kapton」)。該芳香族聚醯亞胺雖然是具有充分的柔軟性以及高耐熱性的芳香族聚醯亞胺,但是會呈現褐色,因而無法用於滿足需具有透光性的替代玻璃用途以及光學用途等。
因而,近年來為了可被用於替代玻璃用途等,而積極開發具有充分透光性的脂環式聚醯亞胺,例如國際公開第2011/099518號(專利文獻1)所公開,具有特定通式所記載的重複單元的聚醯亞胺。該專利文獻1記載的聚醯亞胺具有充分的透光性與高耐熱性。但是上述專利文獻1未特別記載形成薄膜時的厚度方向的相位差。另外,顯示器設備領域中,從改善對比度以及視野角度的觀點出發,希望基板等中採用厚度方向的相位差(rth)的絕對值充分小的薄膜等。
現有技術文獻
專利文獻
專利文獻1:國際公開第2011/099518號
技術實現要素:
[發明要解決的課題]
鑑於上述現有技術所存在的問題課題,本發明的目的在於提供一種聚醯亞胺薄膜,以及使用該聚醯亞胺薄膜的有機電致發光元件,該聚醯亞胺薄膜具有充分高的耐熱性以及透明性,同時厚度方向的相位差(rth)的絕對值為充分小的值。
[解決課題的手段]
為了達成上述目的經本發明者們專心研究後發現,通過使聚醯亞胺薄膜由含有下述通式(1)所表示的重複單元的聚醯亞胺所形成,可獲得一種聚醯亞胺薄膜,其具有充分高的耐熱性以及透明性,同時可使厚度方向的相位差(rth)的絕對值為充分小的值,在波長590nm處測定的厚度方向的相位差(rth)換算為厚度為10μm時的值為-100~100nm(絕對值為100nm以下),從而完成本發明。
具體而言,本發明的聚醯亞胺由含有下述通式(1)所表示的重複單元的聚醯亞胺所形成,且在波長590nm處測定的厚度方向的相位差(rth)換算為厚度為10μm時的值為-100~100nm。
[化1]
式(1)中,r1、r2、r3表示各自獨立的選自氫原子、碳原子數1~10的烷基以及氟原子中的一種,r10表示由下述通式(101)~(108)所表示的基團中的一種,n表示0~12的整數;
[化2]
上述本發明的聚醯亞胺薄膜優選為,在波長590nm處測定的厚度方向的上述相位差(rth)為-50~50nm。
本發明的有機電致發光元件具備上述本發明的聚醯亞胺薄膜。
本發明的有機電致發光顯示器具備上述本發明的聚醯亞胺薄膜。
[發明效果]
根據本發明可以提供一種聚醯亞胺薄膜,以及使用該聚醯亞胺薄膜的有機電致發光元件,該聚醯亞胺薄膜具有充分高的耐熱性以及透明性,同時厚度方向的相位差(rth)的絕對值為充分小的值。
附圖說明
圖1為表示本發明的有機電致發光元件的優選實施方式之一的概略縱剖面圖。
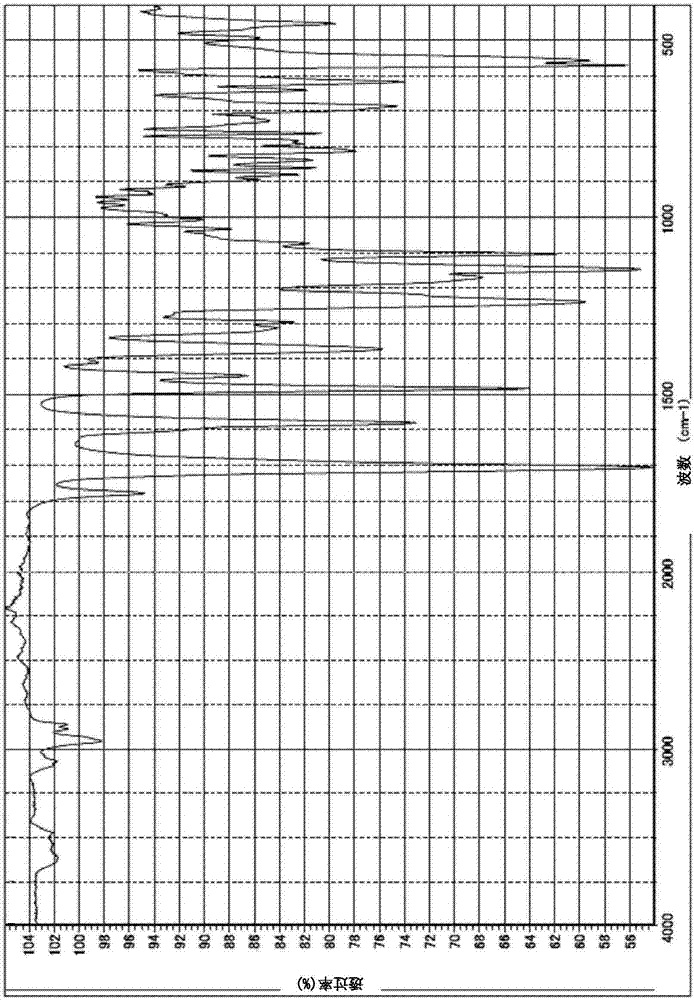
圖2為表示實施例1所得到的聚醯亞胺薄膜的ir光譜圖。
具體實施方式
下面詳細說明本發明的優選實施方式。
[聚醯亞胺薄膜]
本發明的聚醯亞胺薄膜由含有下述通式(1)所表示的重複單元的聚醯亞胺所形成,且在波長590nm處測定的厚度方向的相位差(rth)換算為厚度為10μm時的值為-100~100nm,
[化3]
式(1)中,r1、r2、r3表示各自獨立的選自氫原子、碳原子數1~10的烷基以及氟原子中的一種,r10表示由下述通式(101)~(108)所表示的基團中的一種,n表示0~12的整數,
[化4]
通式(1)中,r1、r2、r3可選用的烷基為,碳原子數1~10的烷基。碳原子數超過10時會降低玻璃化轉變溫度而無法達成充分高的耐熱性。另外,r1、r2、r3可選用的烷基的碳原子數從更容易精製的觀點出發,優選為1~6,較優選為1~5,更優選為1~4,特別優選為1~3。另外,r1、r2、r3可選用的烷基可為直鏈狀或支鏈狀。另外烷基從容易精製的觀點出發,更優選為甲基、乙基。
上述通式(1)中r1、r2、r3從製造聚醯亞胺時可得更高耐熱性的觀點出發,更優選為各自獨立的氫原子或碳原子數1~10的烷基,其中從容易取得原料以及更容易精製的觀點出發,更優選為各自獨立的氫原子、甲基、乙基、n-丙基或異丙基,特別優選為氫原子或甲基。另外,式中的多個r1、r2、r3從容易精製等的觀點出發,特別優選為相同。
另外,上述通式(1)中r10可選用的基團為上述通式(101)~(108)所表示的基團中的至少一種。上述通式(1)中r10為上述通式(101)~(108)所表示的基團時,可得到在波長590nm處測定的厚度方向的相位差(rth)換算為厚度為10μm時的值為-100~100nm(絕對值為100nm以下)的聚醯亞胺薄膜。
從可使rth的絕對值為極小的觀點出發,r10優選為上述通式(101)、(102)、(103)所表示的基團(更優選為上述通式(101)、(102)所表示的基團),從可縮小rth的絕對值的觀點出發,優選為上述通式(104)、(105)、(106)、(107)所表示的基團(更優選為上述通式(104)、(105)所表示的基團)。另外,比較上述通式(101)與通式(107),由於通式(101)中苯環的間位上有來自二胺的結合部位,通式(101)是與通式(107)結構不同的基團,由此可見,在其結構為間位存在來自二胺的鍵合部位的基團中,能夠進一步縮小rth的絕對值,因此與對位為來自二胺的鍵合部位相比較,在通式(101)中,有能夠形成厚度方向的相位差的絕對值為更小的值的薄膜的傾向,從上述觀點出發,r10優選為上述通式(101)、(102)、(103)所表示的基團。
另外,上述通式(1)中n表示0~12的整數。該n值超過上述上限時將難精製。另外,通式(1)中n的數值範圍的上限值從更容易精製的觀點出發,更優選為5,特別優選為3。另外,通式(1)中n的數值範圍的下限值從形成通式(1)所表示的重複單元時所使用的原料化合物的安全性的觀點出發,即更容易製造聚醯亞胺的觀點出發,更優選為1,特別優選為2。因此通式(1)中n特別優選為2~3的整數。
另外聚醯亞胺含有上述通式(1)所表示的重複單元,但優選主要含有上述通式(1)所表示的重複單元。該聚醯亞胺優選為,相對於全部重複單元含有50摩爾%以上的上述通式(1)所表示的重複單元,更優選為含有90~100摩爾%,特別優選含有100摩爾%。通式(1)所表示的重複單元的含有率未達上述下限時會降低作為替代玻璃用途或光學用途等的薄膜用時的特性以及物性。
另外,聚醯亞胺優選為可溶解於低沸點的鑄膜液。由這樣的聚醯亞胺更容易製備形成薄膜。另外,該鑄膜液從溶解性、揮發性、蒸散性、去除性、成膜性、生產性、工業上獲得性、循環性、有無現成設備、價格的觀點出發,優選為沸點200℃以下(優選為20~150℃,更優選為30~120℃,特別優選為40~100℃,最優選為60℃~100℃)的溶劑。另外,作為沸點200℃以下的溶劑,優選為沸點200℃以下的滷系溶劑,更優選為二氯甲烷(氯化甲烯)、三氯甲烷(氯仿)、四氯化碳、二氯乙烷、三氯乙烯、四氯乙烯、四氯乙烷、氯苯、鄰二氯苯,特別優選為二氯甲烷(氯化甲烯)、三氯甲烷(氯仿)。另外,鑄膜液可單獨使用一種或組合使用二種以上。另外,從使所得到的聚醯亞胺相對於鑄膜液的溶解性更高的觀點出發,上述通式(1)中r10更優選為上述通式(101)、(102)、(103)、(104)、(105)、(106)以及(107)所表示的基團中任意一種。
另外,聚醯亞胺優選為亞胺化率(imidizationratio)為90%以上,更優選為95%以上,特別優選為96~100%。亞胺化率未達上述下限時有降低耐熱性、呈現著色、加熱時薄膜發生空隙或膨脹的傾向。
亞胺化率是由下述方法所算出。具體而言,將測定對象的聚醯亞胺溶解於重氯仿等的重溶劑(優選為重氯仿)後,進行1h-nmr測定,再由1h-nmr的圖求出10ppm附近(10ppm±1ppm)的n-h的h與12ppm附近(12ppm±1ppm)的cooh的h的積分值來算出。此時積分比(亞胺化率)的算出如下:首先,將原料化合物的二酸酐以及二胺各自溶解於可溶性重溶劑(dmso-d6等)中製備試料,分別測定1h-nmr光譜,在這些1h-nmr的圖中,求出酸二酐的h位置(化學位移)與積分值以及二胺中的h位置(化學位移)與積分值,再以該酸二酐的h位置與積分值以及二胺中的h位置與積分值作為基準,相對於上述測定對象的聚醯亞胺的1h-nmr的圖中10ppm附近的n-h的h與12ppm附近的cooh的h的積分值,通過相對比較,採用所算出的值。另外,測定該亞胺化率時,使測定1h-nmr光譜的聚醯亞胺量相對於重溶劑(優選為重氯仿)為0.01~5.0質量%的量,原料化合物的酸二酐量以及二胺量分別相對於可溶性重溶劑(dmso-d6等)為0.01~5.0質量%的量。另外,測定該亞胺化率時,聚醯亞胺量、原料化合物的酸二酐量以及二胺量(上述濃度)是在相同濃度下進行測定。另外,上述1h-nmr測定時所採用的測定裝置為nmr測定儀(varian公司制,商品名:unityinova-600)。
另外,上述聚醯亞胺優選為,5%重量減少溫度為400℃以上,更優選為450~550℃。該5%重量減少溫度未達上述下限時有難達成充分耐熱性的傾向,另外,超過上述上限時有難製造具有該特性的聚醯亞胺的傾向。另外,該5%重量減少溫度是在氮氣環境下,一邊使氮氣流動,一邊將掃描溫度設定為30℃~550℃,以10℃/min的條件加熱,所使用的試料的重量減少5%時的溫度來求出。另外,該測定中所使用的測定裝置可為,例如熱重量分析裝置(siinanotechnology制的「tg/dta220」)。
另外,該聚醯亞胺優選玻璃化轉變溫度(tg)為250℃以上,更優選為300~500℃。該玻璃化轉變溫度(tg)未達上述下限時有難達成充分的耐熱性的傾向,另外,超過上述上限時有難製造具有該特性的聚醯亞胺的傾向。另外,該玻璃化轉變溫度(tg),作為測定裝置使用熱機械式分析裝置(rigaku制的商品名「tma8311」),按照測定軟化溫度的同一個方法同時進行測定。另外,測定該玻璃化轉變溫度時優選為,以升溫速度:5℃/分的條件,在氮環境下掃描30℃至550℃的範圍進行測定。
另外,該聚醯亞胺優選為,軟化溫度為250~550℃,優選為350~550℃,更優選為360~510℃。該軟化溫度未達上述下限時會降低耐熱性,例如作為太陽能電池、液晶顯示裝置或有機el顯示裝置的透明電極用的基板使用聚醯亞胺薄膜時,該製品的製造過程中的加熱工序,有難充分抑制該薄膜(基板)的質量劣化(發生破裂等)的傾向,另外,超過上述上限時,製造聚醯亞胺時無法與聚醯胺酸的熱閉環縮合反應同時進行充分的固相聚合反應,故形成薄膜時有形成脆化的薄膜的傾向。
另外,該聚醯亞胺的軟化溫度可按下述方法測定。具體而言,準備作為測定試料用的由長5mm、寬5mm、厚0.013mm(13μm)大小的聚醯亞胺所形成的薄膜,以熱機械式分析裝置(rigaku制的商品名「tma8311」)作為測定裝置,採用氮環境下升溫速度5℃/分的條件,在30℃~550℃的溫度範圍的條件下,通過將透明石英制針(前端直徑:0.5mm)刺入薄膜,可以同時測定玻璃化轉變溫度(tg)。即,所謂的穿透法(penetration)進行測定。另外,該測定可依據jisk7196(1991年)所記載的方法,基於測定數據算出軟化溫度。
另外,這樣形成薄膜的聚醯亞胺優選為,熱分解溫度(td)為450℃以上,更優選為480~600℃。該熱分解溫度(td)未達上述下限時有難達成充分的耐熱性的傾向,另外,超過上述上限時有難製造具有該特性的聚醯亞胺的傾向。另外,該熱分解溫度(td)是由,使用tg/dta220熱重量分析裝置(siinanotechnology制),在氮環境下以升溫速度10℃/min的條件測定熱分解前後的分解曲線的切線的交點的溫度來求出。
另外該聚醯亞胺的數均分子量(mn)以聚苯乙烯進行換算,優選為1000~1000000,更優選為10000~500000。該數均分子量未達上述下限時除了難達成充分耐熱性,且製造時無法由聚合溶劑充分析出,由難有效得到聚醯亞胺的傾向,另外,超過上述上限會增加黏性,而需長時間進行溶解,且需大量溶劑,因而有難加工的傾向。
另外,該形成薄膜的聚醯亞胺的重均分子量(mw)以聚苯乙烯進行換算,優選為1000~5000000。另外,該重均分子量(mw)的數值範圍的下限值優選為5000,更優選為10000,特別優選為20000。另外,重均分子量(mw)的數值範圍的上限值優選為5000000,更優選為500000,特別優選為100000。該重均分子量未達上述下限時不僅有難達成充分耐熱性的傾向,且有製造時無法從聚合溶劑充分析出,難有效得到聚醯亞胺的傾向,另外,超過上述上限時會增加黏性,而需長時間進行溶解,且需大量溶劑,因而有難加工的傾向。
另外該聚醯亞胺的分子量分布(mw/mn)優選為1.1~5.0,更優選為1.5~3.0。該分子量分布未達上述下限時有難製造的傾向,另外,超過上述上限時有不容易得到均勻薄膜的傾向。另外,該聚醯亞胺的分子量(mw或mn)以及分子量分布(mw/mn)可由作為測定裝置的凝膠滲透層析儀(gpc)的測定裝置(脫氧裝置:jasco公司制dg-2080-54,進料泵:jasco公司制pu-2080,接口:jasco公司制lc-netii/adc,柱:shodex公司制gpc管柱kf-806m(×2根),柱箱:jasco公司制860-co,ri檢驗器:jasco公司制ri-2031,柱溫40℃,氯仿溶劑(流速1ml/min.)所測得的數據以聚苯乙烯進行換算來求出。
另外,該聚醯亞胺的線膨脹係數優選為0~100ppm/k,更優選為10~70ppm/k。該線膨脹係數超過上述上限時,將線膨脹係數的範圍為5~20ppm/k的金屬或無機物組合而複合化時有因熱履歷而容易發生剝離的傾向。另外,上述線膨脹係數未達上述下限時具有溶解性降低以及薄膜特性降低的傾向。
該聚醯亞胺的線膨脹係數的測定方法為,形成長76mm、寬52mm、厚13μm大小的聚醯亞胺薄膜後,將該薄膜真空乾燥(120℃下1小時),氮環境下以200℃熱處理1小時後以所得到的乾燥薄膜作為測定用試料,以熱機械式分析裝置(rigaku制的商品名「tma8310」)作為測定裝置,採用氮環境下張力模式(49mn)、升溫速度5℃/分的條件,測定50℃~200℃下上述試料的長度方向的長度變化後,求出50℃~200℃的溫度範圍內每1℃的長度變化的平均值,採用所得到的值。
該聚醯亞胺在不影響本發明效果的範圍內,可含有其它重複單元。該其它重複單元只要是聚醯亞胺可利用的即可,無特別限制。
另外,本發明的聚醯亞胺薄膜是由上述含有通式(1)所表示的重複單元的聚醯亞胺所形成,且在波長590nm處測定的厚度方向的相位差(rth)換算為厚度為10μm時的值為-100~100nm的薄膜。該厚度方向的相位差(rth)超過上述上限時,使用於顯示器設備時有對比度降低以及使視野角度惡化的傾向。另外,該聚醯亞胺薄膜的厚度方向的相位差(rth)換算為厚度為10μm時的值優選為-50~50nm,更優選為-25~25nm。該厚度方向的相位差(rth)超出上述範圍時,使用於顯示器設備時有對比度降低以及使視野角度惡化的傾向。另外,上述的值在範圍內時,使用於顯示器設備時有進一步提高對比度降低抑制以及視野角度改善的效果的傾向。
本發明的聚醯亞胺薄膜的「厚度方向的相位差(rth)」可通過,以axometrics公司制的商品名「axoscan」作為測定裝置,將如後述所測定的聚醯亞胺薄膜的折射率(589nm)輸入上述測定裝置後,以溫度25℃、溼度40%的條件,使用波長590nm的光源測定聚醯亞胺薄膜的厚度方向的相位差,其次基於求得的厚度方向的相位差測定值(由測定裝置自動測定(自動計算)的測定值),換算為薄膜每10μm厚的相位差而求出。另外,測定試料的聚醯亞胺薄膜尺寸可為,大於測定器的測定臺的測光部(直徑:約1cm),無特別限制,優選為長76mm、寬52mm、厚13μm。
另外,測定厚度方向的相位差(rth)時所使用的「上述聚醯亞胺薄膜的折射率(589nm)」可通過,由與形成測定相位差的對象薄膜用的聚醯亞胺相同種類的聚醯亞胺形成未延伸薄膜後,以該未延伸薄膜作為測定試料(另外,測定對象的薄膜為未延伸薄膜時可直接以該薄膜作為測定試料),再以折射率測定裝置(atago株式會社制的商品名「nar-1tsolid」)作為測定裝置,使用589nm的光源,以23℃的溫度條件測定測定試料的平面方向(垂直於厚度方向的方向)相對於589nm光源的折射率而求出。另外,因測定試料是未延伸的,故薄膜平面方向的折射率在平面的任意方向均為一定值,因此通過測定該折射率,可測得該聚醯亞胺固有的折射率(另外,因測定試料是未延伸的,故以平面遲延軸(retardationaxis)方向的折射率為nx,以垂直於遲延軸方向的平面方向的折射率為ny時,nx=ny)。另外,利用未延伸薄膜測定聚醯亞胺固有的折射率(589nm)後,將所得到的測定值用於上述測定厚度方向的相位差(rth)。此時測定試料聚醯亞胺薄膜的尺寸只要是上述折射率測定裝置能夠利用的大小即可,無特別限制,可以是1cm見方(長寬1cm)13μm厚的大小。
另外,該聚醯亞胺薄膜優選為,具有充分高的透明性,優選為全光線透過率為80%以上(更優選為85%以上,特別優選為87%以上)。該全光線透過率容易通過適當選擇聚醯亞胺的種類來達成。另外,該全光線透過率採用的是通過形成長76mm、寬52mm、厚13μm大小的聚醯亞胺薄膜作為測定用試料,然後以日本電色工業株式會社制的商品名「濁度計ndh-5000」作為測定裝置從而測得的值。
該聚醯亞胺薄膜的形態只要是薄膜狀即可,無特別限制,可適當設計為各種形狀(圓盤狀、圓筒狀(將薄膜加工為筒狀)等)。
另外,本發明的聚醯亞胺的厚度無特別限制,優選為1~500μm,更優選為10~200μm。該厚度未達上述下限時會降低強度而有難處理的傾向,另外,超過上述上限時需進行多次塗布,有使加工複雜化的傾向。
另外,本發明的聚醯亞胺薄膜因具有充分高的透明性與耐熱性,故特別適用於例如,可撓性配線基板用薄膜、耐熱絕緣膠帶、電線瓷漆、半導體的保護塗劑、液晶配向膜、有機el用透明導電性薄膜、顯示器的基板材料(tft基板、透明電極基板等的顯示器用基板)、有機el照明用薄膜、可撓性基板薄膜、可撓性有機el用基板薄膜、可撓性透明導電性薄膜、有機薄膜型太陽能電池用透明導電性薄膜、色素增敏型太陽能電池用透明導電性薄膜、可撓性阻氣薄膜、觸控面板用薄膜、可撓性顯示器用正面薄膜、可撓性顯示器用背面薄膜等。另外,本發明的聚醯亞胺薄膜因厚度方向的相位差(rth)的絕對值為充分小的值,因此從改善對比度以及視野角度的觀點出發,特別適用於顯示器用的基板材料(例如tft基板、透明電極基板(有機el用透明導電性薄膜等)等的顯示器用基板)。
製造聚醯亞胺薄膜的方法無特別限制,為了製造由含有上述重複單元的聚醯亞胺所形成的薄膜可採用:使用下述通式(2)所表示的四羧酸二酐與下述通式(301)~(308)所表示的芳香族二胺,適當採用公知的方法(例如國際公開第2011/099518號、國際公開第2014/034760號所記載的聚醯亞胺的製造方法)進行反應來製造聚醯亞胺薄膜的方法。另外,製造聚醯亞胺薄膜的方法優選為,在聚合溶劑存在下,使上述通式(2)所表示的四羧酸二酐與上述通式(301)~(308)所表示的芳香族二胺反應,形成下述通式(4)所表示的聚醯胺酸,然後將含有該聚醯胺酸的聚醯胺酸溶液塗布於基材(例如玻璃基材等)的表面上,接著使該聚醯胺酸醯亞胺化並層疊於上述基板上,由含有上述通式(1)所表示的重複單元的聚醯亞胺所形成的薄膜的方法(a)。
[化5]
[式(2)中,r1、r2、r3、n與上述通式(1)中的r1、r2、r3、n同義。]
[化6]
[化7]
[式(4)中,r1、r2、r3、n與上述通式(1)中的r1、r2、r3、n同義(其優選與上述通式(1)中的r1、r2、r3、n同義),r10與上述通式(1)中的r10同義(其優選也與上述通式(1)中的r10同義)。]
關於通式(2)所表示的四羧酸二酐,通式(2)中的r1、r2、r3為各自獨立的選自氫原子、碳原子數1~10的烷基以及氟原子中的一種,n為0~12的整數。該通式(2)中的r1、r2、r3、n與上述通式(1)中的r1、r2、r3、n相同,其優選與上述通式(1)中的r1、r2、r3、n的優選相同。製造通式(2)所表示的四羧酸二酐的方法無特別限制,可適當採用公知的方法,例如國際公開第2011/099517號所記載的方法以及國際公開第2011/099518號所記載的方法等。
另外,該通式(2)所表示的四羧酸二酐從調整薄膜特性、熱物性、機械物性、光學特性、電特性的觀點出發,優選含有下述通式(5)所表示的化合物(a)與下述通式(6)所表示的化合物(b)中的至少一種,且上述化合物(a)以及(b)的總量為90摩爾%以上。
[化8]
[式(5)中的r1、r2、r3、n與上述通式(2)中的r1、r2、r3、n同義。]
[化9]
[式(6)中的r1、r2、r3、n與上述通式(2)中的r1、r2、r3、n同義。]
通式(5)所表示的化合物(a)是通式(2)所表示的四羧酸二酐的異構體,該通式(2)表示的四羧酸二酐中,2個降冰片烷基為反式排列,並且分別相對於該2個降冰片烷基,環烷酮的羰基為末端構型。另外,通式(6)所表示的化合物(b)是通式(2)所表示的四羧酐二酐的異構體,該通式(2)所表示的四羧酐二酐中,2個降冰片烷基是順式排列,並且分別相對於該2個降冰片烷基,環烷酮的羰基為末端構型。另外,以上述比例含有該異構體的四羧酸二酐的製造方法無特別限制,可適當採用公知的方法,例如可適當採用國際公開第2014/034760號所記載的方法等。
另外,該通式(301)~(308)所表示的芳香族二胺為雙[4-(3-氨基苯氧基)苯基]碸、1,3-雙(3-氨基苯氧基)苯、1,3-雙(4-氨基苯氧基)苯、4,4』-二氨基-2,2-雙(三氟甲基)聯苯、2,2-雙[4-(4-氨基苯氧基)苯基]丙烷、2,2-雙[4-(4-氨基苯氧基)苯基]六氟丙烷、雙[4-(4-氨基苯氧基)苯基]碸、4,4』-雙(4-氨基苯氧基)聯苯。
該芳香族二胺從形成rth的絕對值充分小,且透光率更優良的觀點出發,優選為雙[4-(3-氨基苯氧基)苯基]碸、1,3-雙(3-氨基苯氧基)苯、1,3-雙(4-氨基苯氧基)苯、4,4』-二氨基-2,2-雙(三氟甲基)聯苯(更優選為雙[4-(3-氨基苯氧基)苯基]碸、1,3-雙(3-氨基苯氧基)苯、1,3-雙(4-氨基苯氧基)苯),另外,從形成rth的絕對值充分小,且耐熱性更優良的觀點出發,優選為雙[4-(3-氨基苯氧基)苯基]碸、4,4』-二氨基-2,2-雙(三氟甲基)聯苯、雙[4-(4-氨基苯氧基)苯基]碸(更優選為雙[4-(4-氨基苯氧基)苯基]碸、4,4』-二氨基-2,2-雙(三氟甲基)聯苯、雙[4-(3-氨基苯氧基)苯基]碸)。該芳香族二胺可單獨使用一種或組合使用二種以上。另外,該芳香族二胺可適當使用市售品。
另外,上述方法(a)所使用的上述聚合溶劑優選為,可同時溶解上述通式(2)所表示的四羧酸二酐與上述通式(301)~(308)所表示的芳香族二胺二者的有機溶劑。
該有機溶劑如,n-甲基-2-吡咯烷酮、n,n-二甲基乙醯胺、n,n-二甲基甲醯胺、二甲基亞碸、γ-丁內酯、碳酸亞丙酯、四甲基脲、1,3-二甲基-2-咪唑啉酮、六甲基磷三醯胺、吡啶等的非質子系極性溶劑;m-甲酚、二甲苯酚、苯酚、滷化苯酚等的酚系溶劑;四氫呋喃、二惡烷、溶纖劑、甘醇二甲醚、二甘醇二甲醚等的醚系溶劑;苯、甲苯、二甲苯等的芳香族系溶劑;環戊酮或環己酮等酮系溶劑;乙腈、苯腈等的腈系溶劑等。該聚合溶劑(有機溶劑)可一種單獨或二種以上混合使用。
另外,上述方法(a)中上述通式(2)所表示的四羧酸二酐與上述通式(301)~(308)所表示的芳香族二胺的使用比例優選為,相對於上述通式(301)~(308)所表示的芳香族二胺所具有的1當量氨基,上述通式(2)所表示的四羧酸二酐的酸酐基為0.2~2當量,更優選為0.8~1.2當量。該使用比例未達上述下限時無法有效進行聚合反應,有無法得到高分子量的聚醯胺酸傾向,另外,超過上述上限時,與上述同樣,有無法得到高分子量的聚醯胺酸的傾向。
另外,上述方法(a)中的上述聚合溶劑(有機溶劑)使用量優選為,使上述通式(2)所表示的四羧酸二酐與上述通式(301)~(308)所表示的芳香族二胺的總量,相對於反應溶液全量為0.1~50質量%(更優選為10~30質量%)。該有機溶劑的使用量未達上述下限時,有無法有效得到聚醯胺酸的傾向,另外,超過上述上限時會因高粘度化而有難攪拌的傾向。
另外,上述方法(a)中使上述通式(2)所表示的四羧酸二酐與上述通式(301)~(308)所表示的芳香族二胺反應的方法無特別限制,可適當採用能使四羧酸二酐與芳香族二胺反應的公知的方法,例如可採用大氣壓的條件下,在氮、氦、氬等的惰性環境中,使芳香族二胺類溶解於溶劑後,加入上述通式(2)所表示的四羧酸二酐,其後反應10~48小時的方法。另外,該反應時的溫度條件優選為-20~100℃左右。該反應時間以及反應溫度未達上述下限時,有難充分反應的傾向,另外,超過上述上限時,使聚合物劣化的物質(氧等)的混入概率增大而有降低分子量的傾向。
另外,上述方法(a)中,關於所形成的作為中間物的上述聚醯胺酸,上述通式(4)中的r1、r2、r3、n與上述通式(1)中的r1、r2、r3、n同義,其優選也與上述通式(1)中的r1、r2、r3、n同義。另外,上述通式(4)中的r10與上述通式(1)中的r10同義,其優選也同義。該上述通式(4)中的r10為由上述通式(101)~(108)所表示的基團中所選出的基團。
另外上述方法(a)中所形成的作為中間物的上述聚醯胺酸的固有粘度[η]優選為0.05~3.0dl/g,更優選為0.1~2.0dl/g。該固有粘度[η]小於0.05dl/g時,使用其製造薄膜狀的聚醯亞胺時,有使所得到的薄膜脆化的傾向,另外,超過3.0dl/g時會使粘度過高而降低加工性,例如製造薄膜時難得到均勻的薄膜。
另外,該聚醯胺酸的固有粘度[η]可由下述方法測定。即,首先以n,n-二甲基乙醯胺作為溶劑,將上述聚醯胺酸溶解於該n,n-二甲基乙醯胺中使其濃度為0.5g/dl,得測定試料(溶液)。其次使用上述測定試料,在30℃的溫度條件下使用動粘度計測定上述測定試料的粘度,以所得到的值作為固有粘度[η]用。具體而言,該動粘度計採用離合公司製造的自動粘度測定裝置(商品名「vmc-252」)。
另外,上述方法(a)所使用的基材無特別限制,可對應由目標聚醯亞胺所形成的薄膜的形狀等,適當使用由可用於薄膜成形的公知材料所構成的基材(例如玻璃板或金屬板)。
另外上述方法(a)中,將上述聚醯胺酸的溶液塗布於上述基材上的方法無特別限制,例如可適當採用旋塗法、噴塗法、浸漬塗布法、滴液法、照相凹版印刷法、網版印刷法、凸版印刷法、棒塗法、幕塗法、噴墨法等的公知方法。
上述方法(a)中使上述聚醯胺酸醯亞胺化的方法無特別限制,只要是使聚醯胺酸醯亞胺化的方法即可,無特別限制,可適當採用公知的方法(國際公開第2011/099518號、國際公開第2014/034760號所記載的醯亞胺化的方法等)。該使聚醯胺酸醯亞胺化的方法優選為採用,60~400℃(優選為60~370℃,更優選為150~360℃)的溫度條件下對含有上述通式(4)所表示的重複單元的聚醯胺酸實施加熱處理而醯亞胺化的方法,即所謂的使用「醯亞胺化劑」而醯亞胺化的方法。
另外,該方法(a)可採用,使上述聚醯胺酸醯亞胺化之前,未分離含有上述通式(4)所表示的重複單元的聚醯胺酸下,直接在聚合溶劑(有機溶劑)中使上述通式(2)所表示的四羧酸二酐類與上述通式(301)~308)所表示的芳香族二胺反應所得到的反應液(含有含有上述通式(4)所表示的重複單元的聚醯胺酸的反應液),將上述反應液塗布於基材後,實施乾燥處理以去除溶劑,再通過上述加熱處理而醯亞胺化的方法。該乾燥處理方法的溫度條件優選為0~180℃,更優選為60~150℃。另外,可由上述反應液分離含有上述通式(4)所表示的重複單元的聚醯胺酸再使用,此時聚醯胺酸的分離方法無特別限制,可適當採用能分離聚醯胺酸的公知的方法,例如可採用以再沈澱物方式分離的方法等。
通過該方法(a),可得到層疊於基材上的狀態的聚醯亞胺薄膜。另外,由基材剝離回收所得到的聚醯亞胺薄膜時,該剝離方法無特別限制,可適當採用公知的方法,例如可採用,將基材上層疊聚醯亞胺薄膜的層疊體浸漬於高溫的水(例如80℃以上的水)中,再由基材剝離聚醯亞胺薄膜的方法等。
[有機電致發光元件]
本發明的有機電致發光元件具備上述本發明的聚醯亞胺薄膜。
該有機電致發光元件如,除了具備上述本發明的聚醯亞胺薄膜外,其它構成無特別限制,可適當使用公知的構成物。另外,該有機電致發光元件無特別限制,例如從改善對比度以及視野角度的觀點出發,優選為具備上述本發明的聚醯亞胺薄膜作為透明電極層疊用的基板。
下面參考附圖,簡單說明適用本發明的有機電致發光元件(有機el元件)的實施方式之一。另外,以下說明以及附圖中相同或相當的要素標註相同符號,省略重複說明。
圖1為本發明的有機電致發光元件(有機el元件)的優選實施方式之一的概略縱剖面圖。圖1所示的實施方式的有機el元件1具備聚醯亞胺薄膜11、阻氣層12、透明電極層13、有機層14以及金屬電極層15。
該有機el元件中的聚醯亞胺薄膜11由上述本發明的聚醯亞胺薄膜所形成。該聚醯亞胺薄膜11在本實施方式中,作為有機el元件的基板(透明電極層疊用的基板)。
另外,阻氣層12作為進一步提高防止氣體(包括水蒸氣)透過性能的構成,是適合用於抑制氣體透入元件內部的層。該阻氣層12無特別限制,例如適合使用由sin、sio2、sic、sioxny、tio2、al2o3等無機物所形成的層、超薄板玻璃等。該阻氣層12也可以是在聚醯亞胺薄膜11上適當配置(形成)公知的具有阻氣性的層來層疊得到。
另外,阻氣層12的厚度無特別限制,優選為0.01~5000μm的範圍,更優選為0.1~100μm的範圍。該厚度未達上述下限時有無法得到充分的阻氣性的傾向,另外,超過上述上限時會因厚重化而有可撓性以及柔軟性等特性消失的傾向。
透明電極層13是用作有機el元件的透明電極的層。該透明電極層13的材料只要是能用於有機el元件的透明電極的材料即可,無特別限制,例如可使用氧化銦、氧化鋅、氧化錫以及它們的複合物的銦錫氧化物(ito)、金、鉑、銀、銅,其中從兼具透明性與導電性等的觀點出發,優選為ito。
另外,透明電極層13的厚度優選為20~500nm的範圍。厚度未達上述下限時有使導電性不足的傾向,另外,超過上述上限時會因透明性不足而有使發出的el光無法充分傳到外部的傾向。
另外,阻氣層12與透明電極層13之間,可形成所謂的薄膜電晶體(tft)層。通過以這種方式設置tft層,可以形成具有連接到tft的透明電極的裝置(tft元件)。該tft層的材料(氧化物半導體、非晶矽、多晶矽、有機電晶體等)以及構成無特別限制,可基於公知的tft結構進行適當設計。另外,在聚醯亞胺薄膜11和阻氣層12的層疊體上設置tft層時,也可以使用這些層疊體作為所謂的tft基板。另外,tft層的製造方法無特別限制,可適當採用公知的方法,例如採用低溫多晶矽法、高溫多晶矽法、非晶矽法、氧化物半導體法等的製造方法。
有機層14隻要是能形成有機el元件的材,其構成無特別限制,可適當使用公知的有機el元件所使用的有機層。另外,該有機層14的構成無特別限制,可適當採用公知的構成,例如可以採用由空穴傳輸層、發光層以及電子傳輸層構成的層疊體作為有機層。
該空穴傳輸層的材料可適當使用能形成空穴傳輸層的公知的材料,例如可使用萘二胺(α-npd)、三苯胺、三苯基二胺衍生物(tpd)、聯苯胺、吡唑啉、苯乙烯胺、腙、三苯甲烷、咔唑等的衍生物。
另外,發光層是通過從電極層等注入的電子和空穴的再複合進行發光的層,該發光層的材料無特別限制,可適當使用能形成有機el元件的發光層的公知的材料,例如4,4』-n,n』-二咔唑-聯苯(cbp)上摻雜三苯基吡啶(iii)絡合物(ir(ppy)3)所得到的材料、8-羥基喹啉鋁(alq3、綠色、低分子)、雙(8-羥基)喹哪啶鋁苯氧化物(alq'2oph、藍、低分子)、5,10,15,20-四苯基-21h,23h-卟吩(tpp、紅色、低分子)、聚(9,9-二辛基芴-2,7-二基)(pfo、藍色、高分子)、聚[2-甲氧基-5-(2'-乙基己氧基)-1,4-(1-氰基亞乙烯基)亞苯基](meh-cn-ppv、紅色、高分子)、蒽等的具有螢光性的有機固體所形成的材料等通過施加電壓而發光的公知的材料。
另外電子傳輸層的材料無特別限制,可適當使用能形成電子傳輸層的公知的材料,例如可以使用羥基喹啉鋁絡合物(alq),菲咯啉衍生物,惡二唑衍生物,三唑衍生物,苯基喹喔啉衍生物,噻咯衍生物。
另外,有機層14是由空穴傳輸層、發光層以及電子傳輸層所構成的層疊體時,空穴傳輸層、發光層以及電子傳輸層的各層厚度無特別限制,各自優選為1~50nm的範圍(空穴傳輸層)、5~200nm的範圍(發光層),以及5~200nm的範圍(電子傳輸層)。另外,有機層14的整體厚度優選為20~600nm的範圍。
金屬電極層15是由金屬所構成的電極。該金屬電極的材料可以適當使用功函數較小的材料,無特別限定,例如鋁、mgag、mgin、alli。另外,金屬電極層15的厚度優選為50~500nm的範圍。厚度未達上述下限時有降低導電性的傾向,另外,超過上述上限時有容易剝離以及容易發生斷裂的傾向。
另外,該有機el元件的製造方法無特別限制,例如可採用,在製備如上所述的本發明的聚醯亞胺膜之後,將阻氣層,透明電極,有機層和金屬電極依次層疊在聚醯亞胺膜的表面上。
在聚醯亞胺薄膜11的表面上層疊阻氣層12的方法無特別限制,可適當採用蒸鍍法、濺鍍法等公知的方法,其中從形成周密的膜的觀點出發,優選為採用濺鍍法。另外,在阻氣層12的表面上層疊透明電極層13的方法可為,適當採用蒸鍍法、濺鍍法等公知的方法,其中從形成周密的膜的觀點出發,優選為採用濺鍍法。
另外,在透明電極層13的表面上層疊有機層14的方法也無特別限制,例如,如上所述有機層是由空穴傳輸層、發光層以及電子傳輸層所構成的層疊體時,可以依序在透明電極層13上層疊這些層。另外,層疊有機層14中的各層的方法無特別限制,可適當採用公知的方法,例如可採用蒸鍍法、濺鍍法、塗布法等。這些方法中從充分防止有機層分解、劣化以及變質的觀點出發,優選為採用蒸鍍法。
另外在有機層14上層疊金屬電極層15的方法無特別限制,可適當採用公知的方法,例如可採用蒸鍍法、濺鍍法等。這些方法中從充分防止先前所形成的有機層14分解、劣化以及變質的觀點出發,優選為採用蒸鍍法。
另外,如上述製造有機el元件時,可形成將上述聚醯亞胺薄膜11用作所謂的支撐元件部用的基材的有機el元件,因此通過其rth值可改善對比度以及視野角度,且可形成具有充分高的可撓性的元件。
以上說明的是本發明的有機el元件的優選實施方式之一,但本發明的有機el元件不限定於上述實施方式。例如上述實施方式中,有機層14是由空穴傳輸層、發光層以及電子傳輸層的層疊體所構成的,但有機層的形態無特別限制,可適當採用公知的有機層構成,例如由空穴注入層與發光層的層疊體所構成的有機層;由發光層與電子注入層的層疊體所構成的有機層;由空穴注入層與發光層與電子注入層的層疊體所構成的有機層;或由緩衝層與空穴傳輸層與電子傳輸層的層疊體所構成的有機層等。另外,該有機層的其它形態中,各層的材料無特別限制,可適當使用公知的材料。例如電子注入層的材料可使用苝衍生物等,空穴注入層的材料可使用三苯胺衍生物等,陽極緩衝層的材料可使用銅鈦菁、pedot等。另外,既使是上述實施方式未配置的層,其只要是可用於有機el元件的層即可適當配置,例如從容易將電荷或空穴注入有機層14的觀點出發,透明電極層13上或有機層14上,可設置由氟化鋰(lif)、li2o3等的金屬氟化物、ca、ba、cs等的活性較高的鹼土類金屬、有機絕緣材料等所形成的層。
[有機電致發光顯示器]
本發明的有機電致發光顯示器具備上述本發明的聚醯亞胺薄膜。
該有機電致發光顯示器(有機el顯示器)如,除了具備上述本發明的聚醯亞胺薄膜外,其它構成無特別限制,可適當使用公知的構成。另外,該有機el顯示器優選如,具備作為有機el顯示器所使用的基板(例如層疊透明電極用的基板(透明電極層疊用的基板)、有機el顯示器所使用的阻擋基板、薄膜電晶體基板、密封層基板、負極基板、有機半導體基板、正極基板、直流驅動電路基板、硬塗基板、正面薄膜基板、背面薄膜基板等)用的上述本發明的聚醯亞胺薄膜等。
另外,本發明的有機電致發光顯示器(有機el顯示器)例如從改善對比度以及視野角度的觀點出發,優選為具備上述本發明的聚醯亞胺薄膜作為透明電極層疊用的基板。該透明電極層疊用的基板優選如,有機el顯示器所使用的元件(有機el元件)的透明電極層疊用的基板。因此本發明的有機el顯示器優選具備上述本發明的有機電致發光元件(有機el元件)。
實施例
下面基於實施例以及比較例更具體說明本發明,但本發明不限定於下述實施例。
首先說明各實施例、各比較例所得到的聚醯亞胺薄膜等的特性的評估方法。
<確認分子結構>
通過採用ir測定儀(日本分光株式會社制,商品名:ft/ir-4100)以及nmr測定儀(varian公司制,商品名:unityinova-600),測定ir以及nmr光譜,確認各實施例以及各比較例所得到的化合物的分子結構。
<測定固有粘度[η]>
如上所述,採用離合社製造的自動粘度測定裝置(商品名「vmc-252」),以n,n-二甲基乙醯胺作為溶劑,使用濃度0.5g/dl的測定試料在30℃的溫度條件下,測定各實施例以及各比較例中所得到的作為中間物的聚醯胺酸的固有粘度[η]。
<測定玻璃化轉變溫度(tg)>
各實施例以及各比較例所得到的化合物(形成薄膜的化合物)的玻璃化轉變溫度(tg)通過使用各實施例以及各比較例所製造的聚醯亞胺薄膜,以熱機械式分析裝置(rigaku制的商品名「tma8311」)作為測定裝置,採用與下述軟化溫度的測定方法相同的方法(相同的條件),測定軟化溫度的同時測定得到。
<測定軟化溫度>
各實施例以及各比較例所得到的化合物(形成薄膜的化合物)的軟化溫度(軟化點)通過使用各實施例以及各比較例所製造的聚醯亞胺薄膜,以熱機械式分析裝置(rigaku制的商品名「tma8311」)作為測定裝置,在氮環境下以升溫速度5℃/分、30℃~550℃的溫度範圍(掃描溫度)的條件,將透明石英制針(前端直徑:0.5mm)以500mn壓刺入薄膜進行測定(通過所謂的穿透(刺針)法進行測定)。該測定時除了使用上述測定試料外,是依據jisk7196(1991年)所記載的方法,基於測數據算出軟化溫度。
<測定5%重量減少溫度>
各實施例等所得化合物的5%重量減少溫度(td5%)通過使用各實施例以及各比較例所製造的聚醯亞胺薄膜,以及使用熱重量分析裝置(siinanotechnology制的「tg/dta220」),將掃描溫度設定為30℃~550℃,在氮環境下以氮氣流動的同時以10℃/min的條件進行加熱,測定所使用的試料重量減少5%的溫度所求出。
<測定全光線透過率>
全光線透過率通過使用各實施例以及各比較例所製造的聚醯亞胺薄膜,以日本電色工業株式會社制的商品名「濁度計器ndh-5000」作為測定裝置,依據jisk7361-1(1997年發行)進行測定所求出。
<測定折射率>
各實施例以及各比較例所製造的聚醯亞胺薄膜的折射率(相對於589nm的光線的折射率)通過由同各實施例以及各比較例所採用的方法所製造的聚醯亞胺薄膜(未延伸的薄膜)切出1cm見方(長寬1cm)厚13μm的薄膜作為測定試料,以折射率測定裝置(atago株式會社制的商品名「nar-1tsolid」作為測定裝置,使用589nm的光源,在23℃的溫度條件下,測定相對於589nm的光線的平面方向(與厚度方向垂直的方向)的折射率(聚醯亞胺的固有折射率)所求出。
<厚度方向的相位差(rth)>
厚度方向的相位差(rth)通過直接以各實施例以及各比較例所製造的聚醯亞胺薄膜(長:76mm、寬:52mm、厚:13μm)作為測定試料,以axometrics公司制的商品名「axoscan」作為測定裝置,輸入各聚醯亞胺薄膜的折射率(通過上述測定折射率所求出的薄膜相對於589nm的光線的折射率)的值後,在溫度25℃、溼度40%的條件下,使用波長590nm的光線測定厚度方向的相位差,其次使用所求出的厚度方向的相位差的測定值(由測定裝置自動測定的測定值),換算為每10μm厚薄膜的相位差所求出。
(實施例1)
<準備四羧酸二酐的工序>
依據國際公開第2011/099518號的合成例1、實施例1以及實施例2所記載的方法,準備下述通式(7)所表示的四羧酸二酐(降冰片烷-2-螺-α-環戊酮-α』-螺-2」-降冰片烷-5,5」,6,6」-四羧酸二酐)。
[化10]
<聚醯胺酸的製備工序中>
首先以熱槍加熱30ml的三口燒瓶使其充分乾燥。其次以氮取代充分乾燥後的上述三口燒瓶內的環境氣體,使上述三口燒瓶內成為氮環境。其次將雙[4-(3-氨基苯氧基)苯基]碸0.3892g(0.90mmol,和歌山精化工業株式會社制:baps-m,上述通式(301)所表示的芳香族二胺)加入上述三口燒瓶內,再加入n,n-二甲基乙醯胺2.7g,通過攪拌使芳香族二胺化合物(baps-m)溶解於上述n,n-二甲基乙醯胺得溶解液。
其次在氮環境下將上述通式(7)所表示的化合物0.3459g(0.90mmol)加入含有上述溶解液的三口燒瓶內,氮環境下以室溫(25℃)攪拌12小時得反應液。如此可在反應液中形成聚醯胺酸。
另外,使用部分該反應液(聚醯胺酸的n,n-二甲基乙醯胺溶液:聚醯胺溶液),製備使聚醯胺酸的濃度為0.5g/dl的n,n-二甲基乙醯胺溶液,如上所述測定反應中間物的聚醯胺酸的固有粘度[η],結果聚醯胺酸的固有粘度[η]為0.22dl/g。
<由聚醯亞胺形成薄膜的製備工序>
準備作為玻璃基板用的大型玻璃載片(松浪硝子工業株式會社制的商品名「s9213」,長76mm、寬52mm、厚1.3mm),將上述所得到的反應液(聚醯胺酸溶液)旋塗於上述玻璃基板上,使上述玻璃基板上形成加熱硬化後的厚度為13μm的塗膜。其後將形成上述塗膜的玻璃基板置於60℃的熱板上靜置2小時,由上述塗膜蒸發去除溶劑(溶劑去除處理)。
實施該溶劑去除處理後,將形成上述塗膜的玻璃基板投入氮以3l/分的流量流動的惰性爐內,氮環境下以25℃的溫度條件靜置0.5小時後,以135℃的溫度條件加熱0.5小時,再以350℃的溫度條件(最終加熱溫度)加熱1小時,使上述塗膜硬化,得到在上述玻璃基板上塗覆由聚醯亞胺所形成的薄膜(聚醯亞胺薄膜)的聚醯亞胺塗覆玻璃。
其次將所得到的聚醯亞胺塗覆玻璃浸漬於90℃的熱水中,由上述玻璃基板剝離聚醯亞胺薄膜,得到聚醯亞胺薄膜(長76mm、寬52mm、厚13μm大小的薄膜)。
另外,為了確認形成得到到聚醯亞胺薄膜的化合物的分子結構,使用ir測定儀(日本分光株式會社制,商品名:ft/ir-4100)測定ir光譜。該測定結果所得到的ir光譜如圖2所示。由圖2所得到的結果得知,構成實施例1所形成的薄膜的化合物中,在1706cm-1觀察到醯胺羰基團的c=o伸縮振動。基於該結果確認的分子結構為,所得到的薄膜確實是由聚醯亞胺所形成的薄膜。另外有關所得到的薄膜,如上述測定聚醯亞胺的平面方向相對於589nm的光線的折射率,結果折射率為1.6120。
另外,所得到的聚醯亞胺從所使用的單體種類,相對於全部重複單元,上述通式(1)所表示的重複單元(式中r1、r2、r3均為氫原子,n為2,r10為上述通式(101)所表示的基團的重複單元)的含有率為100摩爾%的聚醯亞胺。另外,有關所得到的聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(實施例2)
聚醯胺酸的製備工序中,除了以1,3-雙(3-氨基苯氧基)苯0.2631g(0.90mmol,東京化成株式會社制1,3,3-bab,上述通式(302)所表示的芳香族二胺)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用,另外由聚醯亞胺形成薄膜的製備工序中,將惰性爐內的上述最終加熱溫度由350℃變更為300℃以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,所得到的聚醯亞胺從所使用的單體種類,相對於全部重複單元,上述通式(1)所表示的重複單元(式中r1、r2、r3均為氫原子,n為2,r10為上述通式(102)所表示的基團的重複單元)的含有率為100摩爾%的聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外有關該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(實施例3)
聚醯胺酸的製備工序中,除了以1,3-雙(4-氨基苯氧基)苯0.2631g(0.90mmol,和歌山精化工業株式會社制1,3,4-bab,上述通式(303)所表示的芳香族二胺)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用,另外有關由聚醯亞胺形成薄膜的製備工序中,將惰性爐內的上述最終加熱溫度由350℃變更為360℃以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,所得到的聚醯亞胺從所使用的單體種類,相對於全部重複單元,上述通式(1)所表示的重複單元(式中r1、r2、r3均為氫原子,n為2,r10為上述通式(103)所表示的基團的重複單元)的含有率為100摩爾%的聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外有關該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(實施例4)
聚醯胺酸的製備工序中,除了以4,4』-二氨基-2,2』-雙(三氟甲基)聯苯0.2882g(0.90mmol,東京化成株式會社制6fda,上述通式(304)所表示的芳香族二胺)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用,同時以氮環境下以60℃攪拌12小時取代氮環境下以室溫25℃)攪拌12小時,得反應液以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,所得到的聚醯亞胺從所使用的單體種類,相對於全部重複單元,上述通式(1)所表示的重複單元(式中r1、r2、r3均為氫原子,n為2,r10為上述通式(104)所表示的基團的重複單元)的含有率為100摩爾%的聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1。另外關於該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(實施例5)
聚醯胺酸的製備工序中,除了以2,2-雙[4-(4-氨基苯氧基)苯基]丙烷0.3695g(0.90mmol,東京化成株式會社制bapp,上述通式(305)所表示的芳香族二胺)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,所得到的聚醯亞胺從所使用的單體種類,相對於全部重複單元,上述通式(1)所表示的重複單元(式中r1、r2、r3均為氫原子,n為2,r10為上述通式(105)所表示的基團的重複單元)的含有率為100摩爾%的聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外關於該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(實施例6)
聚醯胺酸的製備工序中,除了以2,2-雙[4-(4-氨基苯氧基)苯基]六氟丙烷0.4666g(0.90mmol,東京化成株式會社制bapf,上述通式(306)所表示的芳香族二胺)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用,另外由聚醯亞胺形成薄膜的製備工序中,除了使惰性爐內的上述最終加熱溫度由350℃變更為300℃以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,所得到的聚醯亞胺從所使用的單體種類,相對於全部重複單元,上述通式(1)所表示的重複單元(式中r1、r2、r3均為氫原子,n為2,r10為上述通式(106)所表示的基團的重複單元)的含有率為100摩爾%的聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外關於該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(實施例7)
聚醯胺酸的製備工序中,除了以雙[4-(4-氨基苯氧基)苯基]碸0.3892g(0.90mmol,和歌山精化工業株式會社制baps,上述通式(307)所表示的芳香族二胺)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,所得到的聚醯亞胺從所使用的單體種類,相對於全部重複單元,上述通式(1)所表示的重複單元(式中r1、r2、r3均為氫原子,n為2,r10為上述通式(107)所表示的基團的重複單元)的含有率為100摩爾%的聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外有關該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(實施例8)
聚醯胺酸的製備工序中,除了以4,4』-雙(4-氨基苯氧基)聯苯0.3316g(0.90mmol,日本純良製品株式會社制:apbp,上述通式(308)所表示的芳香族二胺)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,所得到的聚醯亞胺為,
因所使用的單體種類,相對於全部重複單元,上述通式(1)所表示的重複單元(式中r1、r2、r3均為氫原子,n為2,r10為上述通式(108)所表示的基團的重複單元)的含有率為100摩爾%的聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外有關該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(比較例1)
聚醯胺酸的製備工序中,除了以下述通式(8)所表示的1,4』-雙(4-氨基苯氧基)苯0.2631g(0.90mmol,東京化成株式會社制:1,4,4-bab)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。
[化11]
另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外關於該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(比較例2)
聚醯胺酸的製備工序中,除了以下述通式(9)所表示的4,4』-二氨基二苯基醚0.1802g(0.90mmol,東京化成株式會社制:4,4』-dde)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。
[化12]
另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外關於該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(比較例3)
聚醯胺酸的製備工序中,除了以下述通式(10)所表示的4,4』-二氨基苯甲醯苯胺0.2045g(0.90mmol,日本純良藥品株式會社制:daban)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用,另外由聚醯亞胺形成薄膜的製備工序中,將惰性爐內的上述最終加熱溫度由350℃變更為380℃以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。
[化13]
另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外關於該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(比較例4)
聚醯胺酸的製備工序中,除了以下述通式(11)所表示的n,n』-雙(4-氨基苯基)對苯二甲醯胺0.3117g(0.90mmol,日本純良藥品株式會社制:data)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用,並且以氮環境下以60℃攪拌12小時取代氮環境下以室溫(25℃)攪拌12小時來得到反應液以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。
[化14]
另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外關於該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
(比較例5)
聚醯胺酸的製備工序中,除了以下述通式(12)所表示的4,4』-二氨基-對三聯苯0.2343g(0.90mmol,東京化成株式會社制:datp)取代雙[4-(3-氨基苯氧基)苯基]碸作為芳香族二胺用,並且在由聚醯亞胺形成薄膜的製備工序中,將惰性爐內的上述最終加熱溫度由350℃變更為400℃以外,其餘採用與實施例1相同的方法,得到聚醯亞胺薄膜。
[化15]
另外,與實施例1同樣確認形成得到的薄膜的化合物的分子結構時,結果確認為聚醯亞胺。另外,關於聚醯胺酸的製備工序中得到的聚醯胺酸,固有粘度[η]的測定結果如表1所示。另外關於該聚醯亞胺薄膜的特性評估結果(通過上述的特性評估方法求出tg以及軟化溫度等)如表1所示。
[表1]
表1中、「―」表示沒有數據檢出。
由表1所示的結果可知,本發明的聚醯亞胺薄膜(實施例1~8)均為,每10μm的厚度方向的相位差(rth)為-100~100nm,確認rth的絕對值為充分低的值(較小的值)。另外,本發明的聚醯亞胺薄膜(實施例1~8)均為,軟化溫度為265℃以上,5%重量減少溫度為463℃以上,確認具有充分高的耐熱性。另外,本發明的聚醯亞胺薄膜(實施例1~8)均為,全光線透過率為87%以上,確認具有充分高的透明性。由該結果得知,各實施例中所得到的聚醯亞胺薄膜(本發明的聚醯亞胺薄膜)為具有高耐熱性與充分透明性,且厚度方向的相位差(rth)的絕對值為充分小的值。
相對於此,各比較例中所得到的聚醯亞胺薄膜,每10μm的厚度方向的相位差(rth)的絕對值為超過100nm的值(比較例1:-121nm、比較例2:-136nm、比較例3:-793nm、比較例4:-931nm、比較例5:-1080nm)、rth的絕對值為100nm以下,故不是具有充分小的rth的絕對值。
產業上的利用可能性
如以上說明,根據本發明可以提供一種具有充分高的耐熱性以及透明性,且厚度方向的相位差(rth)的絕對值可為充分小的值的聚醯亞胺薄膜,以及使用該聚醯亞胺薄膜的有機電致發光元件。
本發明的聚醯亞胺薄膜例如具有充分高的透明性與耐熱性,且厚度方向的相位差(rth)的絕對值為充分小,因而特別適合用作例如可撓性配線基板用薄膜、耐熱絕緣膠帶、電線瓷漆、半導體的保護塗覆劑、液晶配向膜、有機el用透明導電性薄膜、有機el照明用薄膜、可撓性基板薄膜、可撓性有機el用基板薄膜、可撓性透明導電性薄膜、有機薄膜型太陽能電池用透明導電性薄膜、色素增敏型太陽能電池用透明導電性薄膜、可撓性阻氣薄膜、觸控面板用薄膜、可撓性顯示器用正面薄膜、可撓性顯示器用背面薄膜、緩衝塗覆膜、覆蓋膜等。
符號說明
1:有機el元件
11:聚醯亞胺薄膜
12:阻氣層
13:透明電極層
14:有機層
15:金屬電極層