環氧烷聚合催化劑和使用其製造聚環氧烷的方法與流程
2023-06-04 15:18:52 3
本發明涉及環氧烷聚合催化劑和使用其製造聚環氧烷的方法,尤其是涉及即使為低溫的製造條件也能夠高效地製造高分子量、低不飽和度且顯示出狹窄的分子量分布的聚環氧烷的新型環氧烷聚合催化劑、以及使用其製造聚環氧烷的方法。
背景技術:
聚環氧烷通過以氫氧化鉀作為催化劑並進行環氧烷的加聚來製造在工業上是已知的。然而,通過該方法製造高分子量的聚環氧烷時,所得聚環氧烷包含大量一元醇,因此存在不飽和度變高的課題。
作為聚環氧烷的製造方法,提出了使用復金屬氰化物絡合物作為催化劑來製造聚環氧烷的方法(例如參照專利文獻1、2)。
此外,作為不飽和度低的聚環氧烷的製造方法,提出了將特定的膦腈鹽用作催化劑來製造聚環氧烷的方法(例如參照專利文獻3~5)。
另外提出了下述方法:使用將含活性氫的化合物與鋁氧烷以含活性氫的化合物中的活性氫與鋁氧烷中的鋁原子的比率達到0.2~1:1(摩爾比)的方式混合而得到的環氧烷聚合催化劑,進行環氧烷的開環聚合,從而製造聚環氧烷(例如參照專利文獻6)。
進而,作為聚環氧烷的製造方法,提出了下述方法:使用將含活性氫的化合物、膦腈化合物、三異丁基鋁以1:1:2的摩爾比混合得到的環氧烷聚合催化劑,在甲苯溶劑中以20℃進行環氧烷的開環聚合,從而製造聚環氧烷(例如參照非專利文獻1)。關於此時的催化活性種,提出了下述機理:首先,通過含活性氫的化合物與膦腈化合物的反應,進行含活性氫的化合物的脫質子化反應,其後,三異丁基鋁發生作用,經脫質子化的含活性氫的化合物在鋁上移動,從而成為催化活性種。
現有技術文獻
專利文獻
專利文獻1:美國專利第5235114號說明書
專利文獻2:日本特開平4-59825號公報
專利文獻3:日本特許第3497054號說明書(日本特開平10-77289號公報)
專利文獻4:日本特許第3905638號說明書(日本特開平11-106500號公報)
專利文獻5:日本特許第5663856號說明書(日本特開2010-150514號公報)
專利文獻6:日本特開2011-122134號公報
非專利文獻
非專利文獻1:Polymer Chemistry、2012、3、1189.
技術實現要素:
發明要解決的問題
專利文獻1、2所述的方法中,所得聚環氧烷的分子量分布較寬,作為環氧烷存在難以應用於環氧乙烷等的課題。
專利文獻3中提出的方法存在環氧烷的轉化率低至9~19%的課題。此外,所得聚環氧烷是分子量為17000000~160000000g/mol的高分子量體與分子量為1700~2300g/mol的低分子量體的混合物,作為聚環氧烷沒有特殊的特徵。
專利文獻4~6所述的方法中,所得聚環氧烷的不飽和度依然較高,尋求進一步降低。此外,若降低聚合溫度,則產生催化活性降低的課題,反之,若提高聚合溫度,則容易產生在產物中的雜質量的指標即不飽和度變高的課題。
非專利文獻1中提出的方法需要大量使用昂貴的膦腈化合物,作為在工業上有效製造聚環氧烷的方法存在課題。此外,作為所得聚環氧烷也沒有特殊的特徵。
若聚環氧烷的不飽和度變高,則製成聚氨酯樹脂時的交聯密度降低、儲能模量降低,因此存在滯後損耗、壓縮永久形變等物性降低的課題。此外,若聚環氧烷的分子量分布變寬,則存在製成聚氨酯樹脂時的成形性變差的課題。此外,若聚環氧烷中的低分子量成分變多,則製成聚氨酯樹脂時的交聯密度降低、儲能模量降低,因此存在滯後損耗、壓縮永久形變等物性降低的課題。
因而,尋求即使為低溫的製造條件也能夠高效地製造聚環氧烷的環氧烷聚合催化劑和使用其製造聚環氧烷的方法,所述聚環氧烷使用的昂貴膦腈化合物的量少、分子量高、不飽和度低且顯示出狹窄的分子量分布。
用於解決問題的方案
本發明人等為了解決上述課題而進行了深入研究,結果發現:包含路易斯酸和特定膦腈鹽的環氧烷聚合催化劑即使在低溫的製造條件下也能夠高效地製造分子量高、不飽和度低且分子量分布狹窄的聚環氧烷,從而完成了本發明。
即,本發明涉及以下示出的環氧烷聚合催化劑和使用其製造聚環氧烷的方法。
[1]一種環氧烷聚合催化劑,其包含路易斯酸和通式(1)所示的膦腈鹽。
(上述通式(1)中,R1和R2各自獨立地表示氫原子或碳原子數1~20的烴基。此處,可以是R1與R2相互鍵合而形成環結構,也可以是R1彼此相互鍵合或R2彼此相互鍵合而形成環結構。X-表示羥基陰離子、碳原子數1~4的烷氧基陰離子、羧基陰離子、碳原子數2~5的烷基羧基陰離子、或者碳酸氫根陰離子。Y表示碳原子或磷原子,a在Y為碳原子時是2、在Y為磷原子時是3。)。
[2]根據上述[1]所述的環氧烷聚合催化劑,其特徵在於,通式(1)所示的膦腈鹽與路易斯酸的比例為[膦腈鹽]:[路易斯酸]=1:0.002~500(摩爾比)。
[3]根據上述[1]或[2]所述的環氧烷聚合催化劑,其特徵在於,路易斯酸為選自由鋁化合物、鋅化合物和硼化合物組成的組中的至少一種化合物。
[4]根據上述[1]或[2]所述的環氧烷聚合催化劑,其特徵在於,路易斯酸為選自由有機鋁、鋁氧烷和有機鋅組成的組中的至少一種化合物。
[5]根據上述[1]~[4]中任一項所述的環氧烷聚合催化劑,其包含通式(1)所示的膦腈鹽、路易斯酸和含活性氫的化合物。
[上述通式(1)中,R1和R2各自獨立地表示氫原子或碳原子數1~20的烴基。此處,可以是R1與R2相互鍵合而形成環結構,也可以是R1彼此相互鍵合或R2彼此相互鍵合而形成環結構。X-表示羥基陰離子、碳原子數1~4的烷氧基陰離子、羧基陰離子、碳原子數2~5的烷基羧基陰離子、或者碳酸氫根陰離子。Y表示碳原子或磷原子,a在Y為碳原子時是2、在Y為磷原子時是3。]。
[6]根據上述[5]所述的環氧烷聚合催化劑,其特徵在於,含活性氫的化合物為選自由水、羥基化合物、胺化合物、羧酸化合物、硫醇化合物和具有羥基的聚醚多元醇組成的組中的至少1種。
[7]一種上述[5]或[6]所述的環氧烷聚合催化劑的製造方法,其特徵在於,將通式(1)所示的膦腈鹽與含活性氫的化合物混合後,再將它們與路易斯酸混合。
[8]一種聚環氧烷的製造方法,其特徵在於,在上述[1]~[5]中任一項所述的環氧烷聚合催化劑的存在下,進行環氧烷的開環聚合。
[9]一種聚環氧烷,其滿足下述i)~iv)中的全部條件。
i)不飽和度為0.020meq/g以下
ii)Mw/Mn為1.10以下
iii)Mh/f為1000以上
iv)Mh/3以下的分子量的面積比率為2.0%以下
(其中,將以聚苯乙烯作為標準物質由凝膠滲透色譜測定求出的數均分子量記作Mn,將重均分子量記作Mw,將最高峰的分子量記作Mh,將聚環氧烷的官能團數量記作f。)
[10]根據上述[9]所述的聚環氧烷,其特徵在於,由通過JIS K-1557記載的方法算出的聚環氧烷的羥基值和其官能團數量算出的分子量為1000~50000g/mol的範圍。
[11]一種上述[9]或[10]所述的聚環氧烷的製造方法,其特徵在於,在包含膦腈化合物和路易斯酸的環氧烷聚合催化劑的存在下,以含活性氫的化合物作為引發劑,進行環氧烷的開環聚合,並且,相對於前述含活性氫的化合物中的活性氫1摩爾,前述膦腈化合物的用量為0.001~0.1摩爾的範圍。
[12]根據上述[11]所述的聚環氧烷的製造方法,其特徵在於,膦腈化合物為通式(1)所示的膦腈鹽。
(上述通式(1)中,R1和R2各自獨立地表示氫原子或碳原子數1~20的烴基。此處,可以是R1與R2相互鍵合而形成環結構,也可以是R1彼此相互鍵合或R2彼此相互鍵合而形成環結構。X-表示羥基陰離子、碳原子數1~4的烷氧基陰離子、羧基陰離子、碳原子數2~5的烷基羧基陰離子、或者碳酸氫根陰離子。Y表示碳原子或磷原子,a在Y為碳原子時是2、在Y為磷原子時是3。)。
[13]根據上述[11]或[12]所述的聚環氧烷的製造方法,其特徵在於,路易斯酸為選自由鋁化合物、鋅化合物和硼化合物組成的組中的至少一種化合物。
[14]根據上述[11]或[12]所述的聚環氧烷的製造方法,其特徵在於,路易斯酸為選自由有機鋁、鋁氧烷和有機鋅組成的組中的至少一種化合物。
[15]根據上述[11]~[14]中任一項所述的聚環氧烷的製造方法,其特徵在於,膦腈化合物與路易斯酸的比例為[膦腈化合物]:[路易斯酸]=1:0.002~500(摩爾比)。
發明的效果
根據本發明,能夠獲得由副反應導致的雜質生成量少、低溫下的催化活性高的環氧烷聚合催化劑。此外,通過使用本發明的環氧烷聚合催化劑將環氧烷進行聚合,能夠製造分子量高、不飽和度低且分子量分布狹窄的聚環氧烷。
此外,使用分子量高、不飽和度低、分子量分布狹窄、低分子量成分少的本發明的聚環氧烷而得到的聚氨酯樹脂的儲能模量提高,因此可期待滯後損耗、壓縮永久形變等物性的提高。
附圖說明
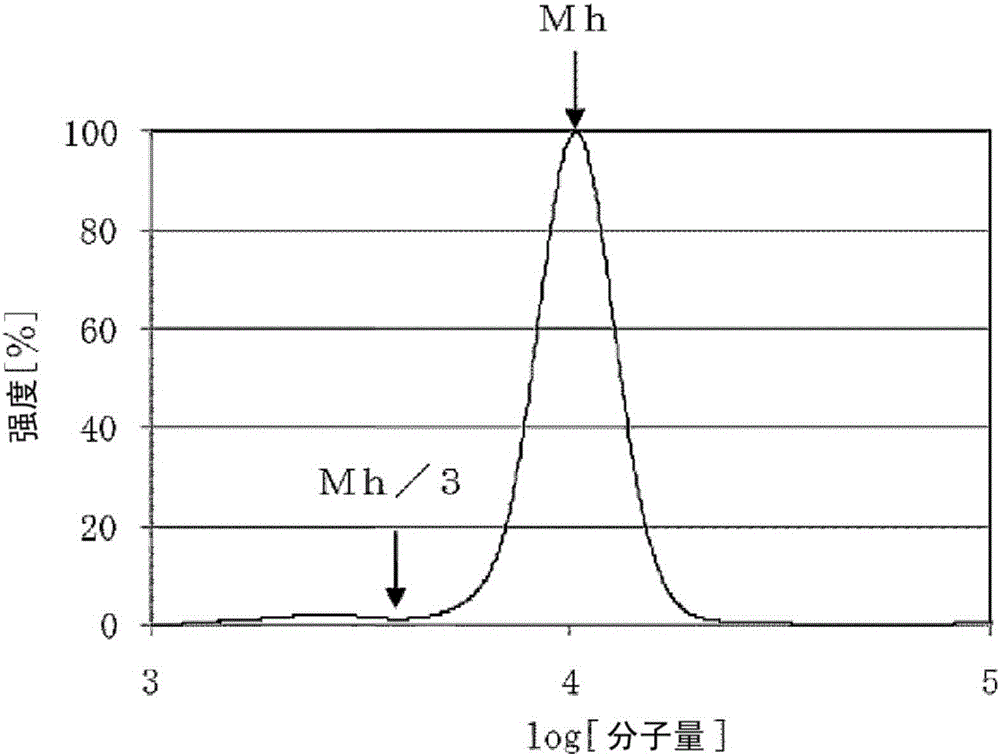
圖1是實施例中的聚環氧烷的低分子量成分的面積比率的計算方法的示意圖。
圖2是實施例中的聚環氧烷的低分子量成分的面積比率的計算方法的示意圖(放大圖)。
具體實施方式
本發明的環氧烷聚合催化劑包含路易斯酸和通式(1)所示的膦腈鹽。
[上述通式(1)中,R1和R2各自獨立地表示氫原子或碳原子數1~20的烴基。此處,可以是R1與R2相互鍵合而形成環結構,也可以是R1彼此相互鍵合或R2彼此相互鍵合而形成環結構。X-表示羥基陰離子、碳原子數1~4的烷氧基陰離子、羧基陰離子、碳原子數2~5的烷基羧基陰離子、或者碳酸氫根陰離子。Y表示碳原子或磷原子,a在Y為碳原子時是2、在Y為磷原子時是3。]。
此處,作為碳原子數1~20的烴基,可列舉出例如甲基、乙基、乙烯基、正丙基、異丙基、環丙基、烯丙基、正丁基、異丁基、叔丁基、環丁基、正戊基、新戊基、環戊基、正己基、環己基、苯基、庚基、環庚基、辛基、環辛基、壬基、環壬基、癸基、環癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十八烷基、十九烷基等。
作為R1與R2相互鍵合而形成環結構的情況,可列舉出例如吡咯烷基、吡咯基、哌啶基、吲哚基、異吲哚基等。
作為R1彼此相互鍵合或R2彼此相互鍵合而形成的環結構,可列舉出例如一個取代基成為亞乙基、亞丙基、亞丁基等亞烷基且與另一個取代基相互鍵合而成的環結構。
並且,這些之中,作為R1和R2,由於形成催化活性優異的環氧烷聚合催化劑,因此,特別優選為甲基、乙基、異丙基。
上述膦腈鹽中的X-為羥基陰離子、碳原子數1~4的烷氧基陰離子、羧基陰離子、碳原子數2~5的烷基羧基陰離子、或者碳酸氫根陰離子。
作為碳原子數1~4的烷氧基陰離子,可列舉出例如甲氧基陰離子、乙氧基陰離子、正丙氧基陰離子、異丙氧基陰離子、正丁氧基陰離子、異丁氧基陰離子、叔丁氧基陰離子等。
作為碳原子數2~5的烷基羧基陰離子,可列舉出例如乙醯氧基陰離子、乙基羧基陰離子、正丙基羧基陰離子、異丙基羧基陰離子、正丁基羧基陰離子、異丁基羧基陰離子、叔丁基羧基陰離子等。
這些之中,作為X-,由於形成催化活性優異的環氧烷聚合催化劑,因此,特別優選為羥基陰離子、碳酸氫根陰離子。
本發明中,Y為碳原子時,膦腈鹽用下述通式(2)表示。
[上述通式(2)中,R1和R2各自獨立地表示氫原子或碳原子數1~20的烴基。此處,可以是R1與R2相互鍵合而形成環結構,也可以是R1彼此相互鍵合或R2彼此相互鍵合而形成環結構。X-表示羥基陰離子、碳原子數1~4的烷氧基陰離子、羧基陰離子、碳原子數2~5的烷基羧基陰離子、或者酸氫根陰離子。]
此外,本發明中,Y為磷原子時,膦腈鹽用下述通式(3)表示。
[上述通式(3)中,R1和R2各自獨立地表示氫原子或碳原子數1~20的烴基。此處,可以是R1與R2相互鍵合而形成環結構,也可以是R1彼此相互鍵合或R2彼此相互鍵合而形成環結構。X-表示羥基陰離子、碳原子數1~4的烷氧基陰離子、羧基陰離子、碳原子數2~5的烷基羧基陰離子、或者酸氫根陰離子。]
本發明中,作為膦腈鹽,具體而言,可例示出四(1,1,3,3-四甲基胍基)氫氧化鏻、四(1,1,3,3-四乙基胍基)氫氧化鏻、四(1,1,3,3-四正丙基胍基)氫氧化鏻、四(1,1,3,3-四異丙基胍基)氫氧化鏻、四(1,1,3,3-四正丁基胍基)氫氧化鏻、四(1,1,3,3-四苯基胍基)氫氧化鏻、四(1,1,3,3-四苄基胍基)氫氧化鏻、四(1,3-二甲基咪唑烷-2-亞氨基)氫氧化鏻、四(1,3-二甲基咪唑烷-2-亞氨基)氫氧化鏻、四(1,1,3,3-四甲基胍基)碳酸氫鏻、四(1,1,3,3-四乙基胍基)碳酸氫鏻、四(1,1,3,3-四正丙基胍基)碳酸氫鏻、四(1,1,3,3-四異丙基胍基)碳酸氫鏻、四(1,1,3,3-四正丁基胍基)碳酸氫鏻、四(1,1,3,3-四苯基胍基)碳酸氫鏻、四(1,1,3,3-四苄基胍基)碳酸氫鏻、四(1,3-二甲基咪唑烷-2-亞氨基)碳酸氫鏻、四(1,3-二甲基咪唑烷-2-亞氨基)碳酸氫鏻等膦腈鹽。
此外,可例示出四[三(二甲基氨基)膦烯氨基]氫氧化鏻、四[三(二乙基氨基)膦烯氨基]氫氧化鏻、四[三(二正丙基氨基)膦烯氨基]氫氧化鏻、四[三(二異丙基氨基)膦烯氨基]氫氧化鏻、四[三(二正丁基氨基)膦烯氨基]氫氧化鏻、四[三(二苯基氨基)膦烯氨基]氫氧化鏻、四[三(1,3-二甲基咪唑烷-2-亞氨基)膦烯氨基]氫氧化鏻、四[三(二甲基氨基)膦烯氨基]碳酸氫鏻、四[三(二乙基氨基)膦烯氨基]碳酸氫鏻、四[三(二正丙基氨基)膦烯氨基]碳酸氫鏻、四[三(二異丙基氨基)膦烯氨基]碳酸氫鏻、四[三(二正丁基氨基)膦烯氨基]碳酸氫鏻、四[三(二苯基氨基)膦烯氨基]碳酸氫鏻、四[三(1,3-二甲基咪唑烷-2-亞氨基)膦烯氨基]碳酸氫鏻等膦腈鹽。
這些之中,由於形成催化性能優異的聚環氧烷製造催化劑,因此,特別優選為四(1,1,3,3-四甲基胍基)氫氧化膦腈、四(1,1,3,3-四甲基胍基)碳酸氫膦腈、四[三(二甲基氨基)膦烯氨基]氫氧化鏻。
本發明中,作為路易斯酸,可列舉出例如鋁化合物、鋅化合物、硼化合物等。
作為鋁化合物,可列舉出例如有機鋁、無機鋁、鋁氧烷等。
作為有機鋁,可列舉出例如三甲基鋁、三乙基鋁、三異丁基鋁、三正己基鋁、三乙氧基鋁、三異丙氧基鋁、三異丁氧基鋁、三苯基鋁、二苯基單異丁基鋁、單苯基二異丁基鋁等。
作為無機鋁,可列舉出氯化鋁、氫氧化鋁、氧化鋁等。
作為鋁氧烷,可列舉出例如下式所示的化合物。
(-AlR3-O-)n
(式中,R3表示碳原子數1~20的烴基。n表示2以上的整數。)
此處,作為碳原子數1~20的烴基,可列舉出例如甲基、乙基、乙烯基、正丙基、異丙基、環丙基、烯丙基、正丁基、異丁基、叔丁基、環丁基、正戊基、新戊基、環戊基、正己基、環己基、苯基、庚基、環庚基、辛基、環辛基、壬基、環壬基、癸基、環癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十八烷基、十九烷基等。這些之中,作為R3,由於形成容易獲取且顯示出高環氧烷轉化率的環氧烷聚合催化劑,因此優選為甲基、異丁基。
作為鋁氧烷的具體例,可列舉出甲基鋁氧烷、異丁基鋁氧烷、甲基異丁基鋁氧烷等。此外,作為鋁氧烷的市售品,可列舉出例如(商品名)MMAO-3A(Tosoh Finechem Corporation制)、(商品名)TMAO-340系列(Tosoh Finechem Corporation制)、(商品名)TMAO-200系列(Tosoh Finechem Corporation制)、(商品名)PBAO(Tosoh Finechem Corporation制)、(商品名)Solid-MAO(Tosoh Finechem Corporation制)等。
作為鋅化合物,可列舉出例如二甲基鋅、二乙基鋅、二苯基鋅等有機鋅;氯化鋅、氧化鋅等無機鋅。
作為硼化合物,可列舉出三乙基硼烷、三甲氧基硼烷、三乙氧基硼烷、三異丙氧基硼烷、三苯基硼烷、三(五氟苯基)硼烷、三氟硼烷等。
並且,這些之中,由於形成催化性能優異的環氧烷聚合催化劑,因此優選為有機鋁、鋁氧烷、有機鋅,特別優選為有機鋁。
此外,這些之中,由於形成容易獲取且催化活性優異的環氧烷聚合催化劑,因此,特別優選為三異丁基鋁、三異丙氧基鋁、二乙基鋅。
本發明的環氧烷聚合催化劑中的膦腈鹽與路易斯酸的比例只要表現出作為環氧烷聚合催化劑的作用就是任意的,沒有特別限定。這些之中,尤其是形成催化活性優異、環氧烷的轉化率高的聚合催化劑,因此優選為[膦腈鹽]:[路易斯酸]=1:0.002~500(摩爾比)。
作為本發明的環氧烷聚合催化劑,由於能夠高效地製造分子量更高、不飽和度更低、且顯示出狹窄分子量分布的聚環氧烷,形成催化活性優異的聚合催化劑,因此,進一步特別優選使用含活性氫的化合物。作為此時的含活性氫的化合物,可列舉出例如水、羥基化合物、胺化合物、羧酸化合物、硫醇化合物、具有羥基的聚醚多元醇等。
作為羥基化合物,可列舉出例如乙二醇、二乙二醇、丙二醇、二丙二醇、1,3-丙二醇、1,3-丁二醇、1,4-丁二醇、1,6-己二醇、甘油、三羥甲基丙烷、己三醇、季戊四醇、二甘油、山梨糖醇、蔗糖、葡萄糖、2-萘酚、雙酚等。
作為胺化合物,可列舉出例如乙二胺、N,N』-二甲基乙二胺、哌啶、哌嗪等。
作為羧酸化合物,可列舉出例如苯甲酸、己二酸等。
作為硫醇化合物,可列舉出例如乙二硫醇、丁二硫醇等。
作為具有羥基的聚醚多元醇,可列舉出例如分子量為200~3000的聚醚多元醇等。
並且,這些含活性氫的化合物可以單獨使用,也可以混合多種使用。
此外,使用含活性氫的化合物時,由於能夠形成可高效地製造分子量更高、不飽和度更低、且顯示出狹窄分子量分布的聚環氧烷且催化活性優異的聚合催化劑,因此,相對於含活性氫的化合物中的活性氫1摩爾,上述膦腈鹽優選為0.001~10摩爾、特別優選為0.001~5摩爾。此外,相對於含活性氫的化合物中的活性氫1摩爾,上述路易斯酸優選為0.001~10摩爾、特別優選為0.001~5摩爾。
本發明中,環氧烷聚合催化劑的製備方法只要能夠形成本發明的聚環氧烷,則可以使用任何方法,沒有特別限定。
可列舉出例如將膦腈鹽與路易斯酸進行混合的方法。此時,作為溶劑,可以使用例如苯、甲苯、二甲苯、環己烷、1,2-二氯乙烷、氯苯、二氯苯、1,4-二噁烷、1,2-二甲氧基乙烷等。
此外,在包含膦腈鹽和路易斯酸的環氧烷聚合催化劑的存在下,以含活性氫的化合物作為引發劑來進行環氧烷的開環聚合時,可以使用將膦腈鹽、路易斯酸和含活性氫的化合物同時混合的方法;向這些之中的1種成分中混合其它的2種成分的方法;向這些之中的2種成分中混合其它的1種成分的方法等任意方法。
這些之中,由於能夠製備可高效地製造分子量更高、不飽和度更低、且顯示出狹窄分子量分布的聚環氧烷且催化活性優異的聚合催化劑,因此優選的是,將上述膦腈鹽與含活性氫的化合物混合後,再將它們與路易斯酸混合,從而製備環氧烷聚合催化劑。此時,也可以進行加熱/減壓處理等,作為加熱處理的溫度,可列舉出例如50~150℃、優選為70~130℃,此外,作為減壓處理時的壓力,可列舉出例如50kPa以下、優選為20kPa以下。
本發明的環氧烷聚合催化劑的催化活性優異,因此對於聚環氧烷的製造而言是有用的,可以在本發明的環氧烷聚合催化劑的存在下進行環氧烷的開環聚合。
作為此時的環氧烷,沒有特別限定,可列舉出例如碳原子數2~20的環氧烷。具體而言,可例示出環氧乙烷、環氧丙烷、1,2-環氧丁烷、2,3-環氧丁烷、異環氧丁烷、一氧化丁二烯、環氧戊烷、環氧苯乙烯、環氧環己烯等。
這些之中,由於容易獲取環氧烷且所得聚環氧烷的工業價值高,因此優選為環氧乙烷、環氧丙烷。環氧烷可以單獨使用,也可以混合2種以上使用。混合2種以上使用時,例如可以使第一環氧烷反應後再與第二環氧烷反應,還可以使2種以上的環氧烷同時反應。
本發明的聚環氧烷的製造方法中,聚合壓力可以為0.05~1.0MPa的範圍、優選為0.1~0.6MPa的範圍。本發明的聚環氧烷的製造中,聚合溫度可以為0~130℃的範圍、優選為10~110℃的範圍。
本發明的聚環氧烷的製造方法中,聚合可以在溶劑中進行,或者在無溶劑的條件下進行。作為使用的溶劑,沒有特別限定,可列舉出例如苯、甲苯、二甲苯、環己烷、1,2-二氯乙烷、氯苯、二氯苯、1,4-二噁烷、1,2-二甲氧基乙烷等。
本發明的聚環氧烷的製造方法中,由於成為有效的聚環氧烷的製造方法,因此,作為催化活性,優選顯示出100g/mol·min以上的催化活性、特別優選顯示出200g/mol·min以上的催化活性。
關於通過本發明的製造方法而得到的聚環氧烷,作為由通過JIS K-1557記載的方法算出的聚環氧烷的羥基值和其官能團數量算出的分子量,優選為1000~50000g/mol、特別優選為3000~30000g/mol。
此外,不飽和度優選為0.05meq/g以下、特別優選為0.03meq/g以下。進而,由聚環氧烷的數均分子量(Mn)、重均分子量(Mw)算出的聚環氧烷的分子量分布(Mw/Mn)優選為1.3以下、特別優選為1.1以下。
接著,針對滿足本發明的下述i)~iv)中的全部條件的聚環氧烷進行說明。
i)不飽和度為0.020meq/g以下
ii)Mw/Mn為1.10以下
iii)Mh/f為1000以上
iv)Mh/3以下的分子量的面積比率為2.0%以下。
[其中,將以聚苯乙烯作為標準物質由凝膠滲透色譜測定求出的數均分子量記作「Mn」、將重均分子量記作「Mw」、將最高峰的分子量記作「Mh」、將聚環氧烷的官能團數量記作「f」。]
本發明的聚環氧烷的不飽和度為0.020meq/g以下、優選為0.010meq/g以下。若不飽和度大於0.020meq/g,則製成聚氨酯樹脂時的儲能模量降低,滯後損耗、壓縮永久形變等物性降低,故不優選。
本發明的聚環氧烷的Mw/Mn為1.10以下、優選為1.08以下。若Mw/Mn大於1.10,則製成聚氨酯樹脂時的儲能模量降低、成形性變差,故不優選。
本發明的聚環氧烷的Mh/f為1000以上、優選為1500以上。若Mh/f小於1000,則製成聚氨酯樹脂時的柔軟性等物性變差,故不優選。
本發明的聚環氧烷的Mh/3以下的分子量的面積比率為2.0%以下、優選為1.0%以下。若Mh/3以下的分子量的面積比率大於2.0%,則製成聚氨酯樹脂時的儲能模量降低,滯後損耗、壓縮永久形變等物性降低,故不優選。
關於本發明的聚環氧烷,作為由通過上述JIS K-1557記載的方法算出的聚環氧烷的羥基值和其官能團數量算出的分子量,優選為1000~50000g/mol、特別優選為3000~30000g/mol。
本發明的聚環氧烷例如通過在包含膦腈化合物和路易斯酸的環氧烷聚合催化劑的存在下,以含活性氫的化合物作為引發劑來進行環氧烷的開環聚合,並且,將前述膦腈化合物相對於前述含活性氫的化合物中的活性氫1摩爾的用量設為0.001~0.1摩爾的範圍,從而能夠簡便地製造。
此處,作為膦腈化合物,具體而言,可例示出四(1,1,3,3-四甲基胍基)氫氧化鏻、四(1,1,3,3-四乙基胍基)氫氧化鏻、四(1,1,3,3-四正丙基胍基)氫氧化鏻、四(1,1,3,3-四異丙基胍基)氫氧化鏻、四(1,1,3,3-四正丁基胍基)氫氧化鏻、四(1,1,3,3-四苯基胍基)氫氧化鏻、四(1,1,3,3-四苄基胍基)氫氧化鏻、四(1,3-二甲基咪唑烷-2-亞氨基)氫氧化鏻、四(1,3-二甲基咪唑烷-2-亞氨基)氫氧化鏻、四(1,1,3,3-四甲基胍基)碳酸氫鏻、四(1,1,3,3-四乙基胍基)碳酸氫鏻、四(1,1,3,3-四正丙基胍基)碳酸氫鏻、四(1,1,3,3-四異丙基胍基)碳酸氫鏻、四(1,1,3,3-四正丁基胍基)碳酸氫鏻、四(1,1,3,3-四苯基胍基)碳酸氫鏻、四(1,1,3,3-四苄基胍基)碳酸氫鏻、四(1,3-二甲基咪唑烷-2-亞氨基)碳酸氫鏻、四(1,3-二甲基咪唑烷-2-亞氨基)碳酸氫鏻等膦腈鹽。
此外,可例示出四[三(二甲基氨基)膦烯氨基]氫氧化鏻、四[三(二乙基氨基)膦烯氨基]氫氧化鏻、四[三(二正丙基氨基)膦烯氨基]氫氧化鏻、四[三(二異丙基氨基)膦烯氨基]氫氧化鏻、四[三(二正丁基氨基)膦烯氨基]氫氧化鏻、四[三(二苯基氨基)膦烯氨基]氫氧化鏻、四[三(1,3-二甲基咪唑烷-2-亞氨基)膦烯氨基]氫氧化鏻、四[三(二甲基氨基)膦烯氨基]碳酸氫鏻、四[三(二乙基氨基)膦烯氨基]碳酸氫鏻、四[三(二正丙基氨基)膦烯氨基]碳酸氫鏻、四[三(二異丙基氨基)膦烯氨基]碳酸氫鏻、四[三(二正丁基氨基)膦烯氨基]碳酸氫鏻、四[三(二苯基氨基)膦烯氨基]碳酸氫鏻、四[三(1,3-二甲基咪唑烷-2-亞氨基)膦烯氨基]碳酸氫鏻等膦腈鹽。
此外,可例示出1-叔丁基-4,4,4-三(二甲基氨基)-2,2-雙(三(二甲基氨基)膦烯氨基)-2λ5,4λ5-聯二膦腈。
此處,作為膦腈化合物,優選為上述通式(1)所示的膦腈鹽。
本發明的聚環氧烷由於分子量高、不飽和度低、分子量分布狹窄、低分子量成分少,因此,可期待使用其得到的聚氨酯樹脂的儲能模量提高,滯後損耗、壓縮永久形變等物性提高。
實施例
以下,通過實施例來說明本發明,但本實施例完全不限定本發明。
首先,針對催化活性的計算方法、聚環氧烷的分析方法進行說明。
(1)催化活性(單位:g/mol·min)
將已反應的環氧烷量記作a(單位:g),將使用的膦腈鹽量記作b(單位:mol),將聚合所需的時間記作c(單位:min),通過下式來算出催化活性。
催化活性=a/(b×c)。
(2)聚環氧烷的分子量(單位:g/mol)
通過JIS K-1557記載的方法,測定聚環氧烷的羥基值d(單位:mgKOH/g)。將所得聚環氧烷的官能團數量記作e,通過下式來算出聚環氧烷的分子量。
分子量=(56100/d)×e。
此外,使用凝膠滲透色譜(GPC)(東曹株式會社制、HLC8020),以四氫呋喃作為溶劑,在40℃下進行測定,作為標準物質而使用聚苯乙烯,算出聚環氧烷的數均分子量(Mn)、重均分子量(Mw)、最高峰的分子量(Mh)。
(3)聚環氧烷的低分子量成分的面積比率(單位:%)
計算通過上述方法(2)算出的Mh除以3而得到的分子量(Mh/3)以下、即低分子量成分的面積比率。
(4)聚環氧烷的不飽和度(單位:meq/g)
通過JIS K-1557記載的方法,算出聚環氧烷的不飽和度。
(5)聚環氧烷的分子量分布(單位:無)
根據通過上述方法(2)算出的數均分子量(Mn)、重均分子量(Mw),算出該聚環氧烷的分子量分布(Mw/Mn)。
合成例1(膦腈鹽A的合成)
將附帶攪拌葉片的2升四頸燒瓶中製成氮氣氣氛,添加五氯化磷96g(0.46mol)、脫水甲苯800ml,以20℃進行攪拌。在維持攪拌的條件下,耗費3小時滴加1,1,3,3-四甲基胍345g(2.99mol)後,升溫至100℃,進一步耗費1小時滴加1,1,3,3-四甲基胍107g(0.92mol)。將所得白色的漿料溶液以100℃攪拌14小時後,冷卻至80℃,添加離子交換水250ml,攪拌30分鐘。停止攪拌時,漿料全部溶解而得到二相溶液。進行所得二相溶液的油水分離,回收水相。向所得水相中添加二氯甲烷100ml,進行油水分離,回收二氯甲烷相。將所得二氯甲烷溶液用離子交換水100ml進行清洗。
將所得二氯甲烷溶液轉移至附帶攪拌葉片的2升四頸燒瓶中,添加2-丙醇900g後,在常壓下將溫度提升至80~100℃,去除二氯甲烷。一邊攪拌所得2-丙醇溶液,一邊將內部溫度放冷至60℃後,添加85重量%的氫氧化鉀31g(0.47mol、相對於亞氨基膦腈鹽為1.1mol當量),以60℃反應2小時。將溫度冷卻至25℃,通過過濾而去除所析出的副產鹽,從而以25重量%的濃度、92%的收率得到作為目標的亞氨基膦腈鹽A[上述通式(1)中的R1為甲基、R2為甲基、X-為羥基陰離子、Y為碳原子、a相當於2的膦腈鹽]的2-丙醇溶液860g。
合成例2(膦腈鹽B的合成)
將附帶磁性轉子的100ml舒倫克瓶製成氮氣氣氛,添加四[三(二甲基氨基)膦烯氨基]氯化磷5.7g(7.4mmol、Aldrich公司制)、2-丙醇16ml,以25℃進行攪拌而使其溶解。在維持攪拌的條件下,添加將85重量%的氫氧化鉀0.53g〔8.1mmol、相對於四[三(二甲基氨基)膦烯氨基]氯化鏻為1.1mol當量〕溶解於2-丙醇而得到的溶液。以25℃攪拌5小時後,通過過濾而去除所析出的副產鹽,從而以17重量%的濃度、98%的收率得到作為目標的膦腈鹽B[上述通式(1)中的R1為甲基、R2為甲基、X-為羥基陰離子、Y為磷原子、a相當於3的膦腈鹽]的2-丙醇溶液32.7g。
實施例1
向附帶攪拌葉片的0.2升高壓釜中添加通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液10g(5mmol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下去除2-丙醇。其後,添加三異丁基鋁的1.0mol/l的甲苯溶液10ml(10mmol)並混合,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成20℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷108g,一邊在內部溫度為18~22℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧丙烷和甲苯的去除,從而得到無色無臭的聚環氧烷106g。催化活性為350g/mol·min、所得聚環氧烷的分子量為20000g/mol、不飽和度為0.018meq/g、分子量分布為1.08。將結果示於表1。
實施例2
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)18g(18mmol、活性氫量為54mmol)、以及通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.54g(0.27mmol、相對於活性氫1mol為0.005mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加三異丁基鋁的1.0mol/l的甲苯溶液0.54ml(0.54mmol、相對於活性氫1mol為0.010mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為68~72℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為68~72℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷127g。催化活性為840g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.017meq/g、分子量分布為1.07。將結果示於表1。
實施例3
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)18g(18mmol、活性氫量為54mmol)、以及通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.27g(0.14mmol、相對於活性氫1mol為0.0025mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加三異丁基鋁的1.0mol/l的甲苯溶液0.27ml(0.27mmol、相對於活性氫1mol為0.005mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為68~72℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為68~72℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷126g。催化活性為1100g/mol·min、所得聚環氧烷的分子量為6800g/mol、不飽和度為0.016meq/g、分子量分布為1.06。將結果示於表1。
實施例4
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)18g(18mmol、活性氫量為54mmol)、以及通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.54g(0.27mmol、相對於活性氫1mol為0.005mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加三異丁基鋁的1.0mol/l的甲苯溶液0.54ml(0.54mmol、相對於活性氫1mol為0.010mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成50℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為48~52℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為48~52℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷125g。催化活性為550g/mol·min、所得聚環氧烷的分子量為6800g/mol、不飽和度為0.014meq/g、分子量分布為1.05。將結果示於表1。
實施例5
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)6.0g(6.0mmol、活性氫量為18mmol)、以及通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.36g(0.18mmol、相對於活性氫1mol為0.010mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加三異丁基鋁的1.0mol/l的甲苯溶液0.36ml(0.36mmol、相對於活性氫1mol為0.020mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為68~72℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為68~72℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷112g。催化活性為980g/mol·min、所得聚環氧烷的分子量為18000g/mol、不飽和度為0.022meq/g、分子量分布為1.08。將結果示於表1。
實施例6
使用二乙基鋅的1.0mol/l的己烷溶液0.54ml(0.54mmol、相對於活性氫1mol為0.010mol)來代替三異丁基鋁的1.0mol/l的甲苯溶液0.54ml(0.54mmol、相對於活性氫1mol為0.010mol),除此之外,通過與實施例2相同的方法,進行環氧烷聚合催化劑、聚環氧烷的製造。得到無色無臭的聚環氧烷127g。催化活性為750g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.016meq/g、分子量分布為1.06。將結果示於表1。
[表1]
比較例1
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)6.0g(6.0mmol、活性氫量為18mmol)、以及作為膦腈P4鹼的1-叔丁基-4,4,4-三(二甲基氨基)-2,2-雙(三(二甲基氨基)膦烯氨基)-2λ5,4λ5-聯二膦腈的1.0mol/l的己烷溶液18ml(18mmol、相對於活性氫1mol為1.0mol)。將高壓釜內製成氮氣氣氛後,添加三異丁基鋁的2.0mol/l的甲苯溶液18ml(36mmol、相對於活性氫1mol為2.0mol)並混合,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成20℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷37g,一邊在內部溫度為18~22℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧丙烷的去除,從而得到無色無臭的聚環氧烷42g。催化活性為11g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.016meq/g、分子量分布為1.36。將結果示於表2。
比較例2
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)18g(18mmol、活性氫量為54mmol)、以及通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.54g(0.27mmol、相對於活性氫1mol為0.005mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷,一邊在內部溫度為68~72℃的範圍內發生反應。其結果,在供給36g環氧丙烷的時刻,反應終止。在0.5kPa的減壓下進行殘留環氧丙烷的去除,從而得到無色無臭的聚環氧烷36g。催化活性為46g/mol·min、所得聚環氧烷的分子量為2000g/mol、不飽和度為0.013meq/g、分子量分布為1.05。將結果示於表2。
[表2]
實施例7
向附帶攪拌葉片的0.2升高壓釜中添加通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液10g(5mmol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下去除2-丙醇。其後,添加具有甲基和異丁基的鋁氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液10ml(鋁原子10mmol)並混合,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成35℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷108g,一邊在內部溫度為33~37℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧丙烷和甲苯的去除,從而得到無色無臭的聚環氧烷106g。環氧烷的轉化率為98%、所得聚環氧烷的分子量為19000g/mol、不飽和度為0.016meq/g、分子量分布為1.07。將結果示於表3。
實施例8
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)18g(18mmol、活性氫量為54mmol)、以及通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.54g(0.27mmol、相對於活性氫1mol為0.005mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加具有甲基和異丁基的鋁氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液0.54ml(鋁原子為0.54mmol、相對於活性氫1mol為0.010mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為68~72℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為68~72℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷127g。環氧烷的轉化率為99%、所得聚環氧烷的分子量為7000g/mol、不飽和度為0.016meq/g、分子量分布為1.06。將結果示於表3。
實施例9
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)18g(18mmol、活性氫量為54mmol)、以及通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.27g(0.14mmol、相對於活性氫1mol為0.0025mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加具有甲基和異丁基的鋁氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液0.27ml(鋁原子為0.27mmol、相對於活性氫1mol為0.005mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為68~72℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為68~72℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷126g。環氧烷的轉化率為98%、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.014meq/g、分子量分布為1.05。將結果示於表3。
實施例10
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)18g(18mmol、活性氫量為54mmol)、以及通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.54g(0.27mmol、相對於活性氫1mol為0.005mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加具有甲基和異丁基的鋁氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液0.54ml(鋁原子為0.54mmol、相對於活性氫1mol為0.010mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成50℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為48~52℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為48~52℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷125g。環氧烷的轉化率為97%、所得聚環氧烷的分子量為6700g/mol、不飽和度為0.012meq/g、分子量分布為1.04。將結果示於表3。
實施例11
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)6.0g(6.0mmol、活性氫量為18mmol)、以及通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.36g(0.18mmol、相對於活性氫1mol為0.010mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加具有甲基和異丁基的鋁氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液0.36ml(鋁原子為0.36mmol、相對於活性氫1mol為0.020mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為68~72℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為68~72℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷112g。環氧烷的轉化率為96%、所得聚環氧烷的分子量為18000g/mol、不飽和度為0.021meq/g、分子量分布為1.08。將結果示於表3。
[表3]
比較例3
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)6.0g(6.0mmol、活性氫量為18mmol)、以及具有甲基和異丁基的鋁氧烷(Tosoh Finechem Corporation制、(商品名)MMAO-3A)的1.0mol/l的甲苯溶液36ml(鋁原子36mmol、相對於活性氫1mol為2.0mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成35℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷,一邊在內部溫度為33~37℃的範圍內發生反應。其結果,在供給12g環氧丙烷的時刻,反應終止。在0.5kPa的減壓下進行殘留環氧丙烷的去除,從而得到無色無臭的聚環氧烷9.0g。環氧烷的轉化率為25%、所得聚環氧烷的分子量為1500g/mol、不飽和度為0.012meq/g、分子量分布為1.42。將結果示於表4。
比較例4
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)18g(18mmol、活性氫量為54mmol)、以及通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.54g(0.27mmol、相對於活性氫1mol為0.005mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷,一邊在內部溫度為68~72℃的範圍內發生反應。其結果,在供給36g環氧丙烷的時刻,反應終止。在0.5kPa的減壓下進行殘留環氧丙烷的去除,從而得到無色無臭的聚環氧烷36g。環氧烷的轉化率為50%、所得聚環氧烷的分子量為2000g/mol、不飽和度為0.013meq/g、分子量分布為1.05。將結果示於表4。
[表4]
實施例12
向附帶攪拌葉片的0.2升高壓釜中添加通過合成例2得到的膦腈鹽B的17重量%2-丙醇溶液11g(2.5mmol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下去除2-丙醇。其後,添加三異丁基鋁的1.0mol/l的甲苯溶液7.5ml(7.5mmol)並混合,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成20℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷108g,一邊在內部溫度為68~72℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧丙烷和甲苯的去除,從而得到無色無臭的聚環氧烷106g。催化活性為400g/mol·min、所得聚環氧烷的分子量為20000g/mol、不飽和度為0.008meq/g、分子量分布為1.08。將結果示於表5。
實施例13
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇[三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g]18g(18mmol、活性氫量為54mmol)、以及通過合成例2得到的膦腈鹽B的17重量%2-丙醇溶液2.0g(0.45mmol、相對於活性氫1mol為0.008mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加三異丁基鋁的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相對於活性氫1mol為0.025mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為68~72℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為108~112℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷127g。催化活性為1040g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.007meq/g、分子量分布為1.07。將結果示於表5。
實施例14
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇[三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g]18g(18mmol、活性氫量為54mmol)、以及通過合成例2得到的膦腈鹽B的17重量%2-丙醇溶液1.0g(0.23mmol、相對於活性氫1mol為0.004mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加三異丁基鋁的1.0mol/l的甲苯溶液0.68ml(0.68mmol、相對於活性氫1mol為0.013mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為68~72℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為108~112℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷126g。催化活性為1300g/mol·min、所得聚環氧烷的分子量為6800g/mol、不飽和度為0.005meq/g、分子量分布為1.06。將結果示於表5。
實施例15
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇[三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g]18g(18mmol、活性氫量為54mmol)、以及通過合成例2得到的膦腈鹽B的17重量%2-丙醇溶液1.2g(0.27mmol、相對於活性氫1mol為0.005mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加三異丁基鋁的1.0mol/l的甲苯溶液0.54ml(0.54mmol、相對於活性氫1mol為0.010mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為78~82℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為108~112℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷125g。催化活性為650g/mol·min、所得聚環氧烷的分子量為6800g/mol、不飽和度為0.007meq/g、分子量分布為1.05。將結果示於表5。
實施例16
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇(三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g)6.0g(6.0mmol、活性氫量為18mmol)、以及通過合成例2得到的膦腈鹽B的25重量%2-丙醇溶液0.54g(0.18mmol、相對於活性氫1mol為0.010mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理。其後,添加三異丁基鋁的1.0mol/l的甲苯溶液0.36ml(0.36mmol、相對於活性氫1mol為0.020mol),將內部溫度製成80℃,進行0.5kPa的減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊在內部溫度為68~72℃的範圍內發生反應。接著,在0.5kPa的減壓下進行殘留環氧丙烷的去除,然後一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊在內部溫度為108~112℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷112g。催化活性為980g/mol·min、所得聚環氧烷的分子量為18000g/mol、不飽和度為0.008meq/g、分子量分布為1.08。將結果示於表5。
實施例17
使用三異丙氧基鋁0.15g(0.75mmol、相對於活性氫1mol為0.025mol)來代替三異丁基鋁的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相對於活性氫1mol為0.008mol),除此之外,通過與實施例13相同的方法,進行環氧烷聚合催化劑、聚環氧烷的製造。從而得到無色無臭的聚環氧烷127g。催化活性為750g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.007meq/g、分子量分布為1.05。將結果示於表5。
[表5]
實施例18
使用三苯基鋁1.0M溶液1.35ml(1.35mmol、相對於活性氫1mol為0.025mol)來代替三異丁基鋁的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相對於活性氫1mol為0.025mol),除此之外,通過與實施例13相同的方法,進行環氧烷聚合催化劑、聚環氧烷的製造。從而得到無色無臭的聚環氧烷127g。催化活性為680g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.006meq/g、分子量分布為1.06。將結果示於表6。
實施例19
使用氯化鋁0.18g(1.35mmol、相對於活性氫1mol為0.025mol)來代替三異丁基鋁的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相對於活性氫1mol為0.025mol),除此之外,通過與實施例13相同的方法,進行環氧烷聚合催化劑、聚環氧烷的製造。從而得到無色無臭的聚環氧烷126g。催化活性為650g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.006meq/g、分子量分布為1.07。將結果示於表6。
實施例20
使用純異丁基鋁氧烷1.0M己烷溶液1.35ml(1.35mmol、相對於活性氫1mol為0.025mol)來代替三異丁基鋁的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相對於活性氫1mol為0.025mol),除此之外,通過與實施例13相同的方法,進行環氧烷聚合催化劑、聚環氧烷的製造。從而得到無色無臭的聚環氧烷107g。催化活性為350g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.006meq/g、分子量分布為1.06。將結果示於表6。
實施例21
除了將高壓釜的內部溫度從70℃變更為120℃之外,通過與實施例13相同的方法,進行環氧烷聚合催化劑、聚環氧烷的製造。從而得到無色無臭的聚環氧烷127g。催化活性為850g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.006meq/g、分子量分布為1.06。將結果示於表6。
實施例22
使用二乙基鋅的1.0mol/l的己烷溶液1.35ml(1.35mmol、相對於活性氫1mol為0.025mol)來代替三異丁基鋁的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相對於活性氫1mol為0.025mol),除此之外,通過與實施例13相同的方法,進行環氧烷聚合催化劑、聚環氧烷的製造。從而得到無色無臭的聚環氧烷127g。催化活性為750g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.010meq/g、分子量分布為1.07。將結果示於表6。
[表6]
比較例5
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇[三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g]6.0g(6.0mmol、活性氫量為18mmol)、以及作為膦腈P4鹼的1-叔丁基-4,4,4-三(二甲基氨基)-2,2-雙(三(二甲基氨基)膦烯氨基)-2λ5,4λ5-聯二膦腈的1.0mol/l的己烷溶液18ml(18mmol、相對於活性氫1mol為1.0mol)。將高壓釜內製成氮氣氣氛後,添加三異丁基鋁的2.0mol/l的甲苯溶液18ml(36mmol、相對於活性氫1mol為2.0mol)並混合,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成20℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷37g,一邊在內部溫度為18~22℃的範圍內發生反應。在0.5kPa的減壓下進行殘留環氧丙烷的去除,從而得到無色無臭的聚環氧烷42g。催化活性為11g/mol·min、所得聚環氧烷的分子量為6900g/mol、不飽和度為0.016meq/g、分子量分布為1.36。將結果示於表7。
比較例6
除了將膦腈鹽0.45mmol設為KOH 0.45mmol之外,利用與實施例13相同的方法,進行環氧烷聚合催化劑、聚環氧烷的製造。從而得到無色無臭的聚環氧烷36g。催化活性為61g/mol·min、所得聚環氧烷的分子量為2000g/mol、不飽和度為0.030meq/g、分子量分布為1.05。將結果示於表7。
比較例7
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇[三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g]18g(18mmol、活性氫量為54mmol)、以及通過合成例2得到的膦腈鹽B的17重量%2-丙醇溶液2.0g(0.45mmol、相對於活性氫1mol為0.008mol)。將高壓釜內製成氮氣氣氛後,將內部溫度製成80℃,在0.5kPa的減壓下進行脫水處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷,一邊在內部溫度為88~92℃的範圍內發生反應。其結果,供給了110g環氧丙烷。在0.5kPa的減壓下進行殘留環氧丙烷的去除,從而得到無色無臭的聚環氧烷125g。催化活性為460g/mol·min、所得聚環氧烷的分子量為7000g/mol、不飽和度為0.024meq/g、分子量分布為1.06。將結果示於表3。
比較例8
向附帶攪拌葉片的0.2升高壓釜中添加作為含活性氫的化合物的具有3個羥基且分子量為1000的聚醚多元醇[三洋化成工業株式會社制、(商品名)SANNIX GP1000;羥基值為160mgKOH/g]18g(18mmol、活性氫量為54mmol)、三異丁基鋁的1.0mol/l的甲苯溶液1.35ml(1.35mmol、相對於活性氫1mol為0.025mol),將內部溫度製成80℃,進行0.5kPa的減壓處理。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,使環氧丙烷在反應壓力0.3MPa以下、內部溫度68~72℃的範圍內發生反應。然而,在70℃的反應溫度下,反應壓力達到0.3MPa後,完全未觀察到壓力的降低。以70℃持續反應5小時後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。回收10g作為原料的聚醚多元醇。三異丁基鋁單獨存在時,完全顯示不出環氧丙烷的聚合活性。將結果示於表7。
[表7]
實施例23
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)18g(活性氫量為54mmol)、通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.90g(0.45mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加三異丁基鋁(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷127g。催化活性為600g/mol·min、所得聚環氧烷的不飽和度為0.006meq/g、分子量分布為1.06、Mh/f為3700g/mol、Mh/3以下的低分子量成分的面積比率為0.1%。
實施例24
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)18g(活性氫量為54mmol)、通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.54g(0.27mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加三異丁基鋁(TIBAL)的1.0mol/l甲苯溶液0.54ml(0.54mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷126g。催化活性為600g/mol·min、所得聚環氧烷的不飽和度為0.005meq/g、分子量分布為1.05、Mh/f為3600g/mol、Mh/3以下的低分子量成分的面積比率為0.1%。
實施例25
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)18g(活性氫量為54mmol)、通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.90g(0.45mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加三異丁基鋁(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成70℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷125g。催化活性為400g/mol·min、所得聚環氧烷的不飽和度為0.005meq/g、分子量分布為1.05、Mh/f為3600g/mol、Mh/3以下的低分子量成分的面積比率為0.1%。
實施例26
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)18g(活性氫量為54mmol)、通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.90g(0.45mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加三異丁基鋁(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成120℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷128g。催化活性為1500g/mol·min、所得聚環氧烷的不飽和度為0.008meq/g、分子量分布為1.06、Mh/f為3700g/mol、Mh/3以下的低分子量成分的面積比率為0.2%。
實施例27
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)9g(活性氫量27mmol)、通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.45g(0.23mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加三異丁基鋁(TIBAL)的1.0mol/l甲苯溶液0.68ml(0.68mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷117g。催化活性為500g/mol·min、所得聚環氧烷的不飽和度為0.008meq/g、分子量分布為1.06、Mh/f為7000g/mol、Mh/3以下的低分子量成分的面積比率為0.2%。
實施例28
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.90g(0.45mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加三異丁基鋁(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷109g。催化活性為500g/mol·min、所得聚環氧烷的不飽和度為0.008meq/g、分子量分布為1.06、Mh/f為7000g/mol、Mh/3以下的低分子量成分的面積比率為0.2%。
實施例29
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制SANNIX GP1000)18g(活性氫量為54mmol)、通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.90g(0.45mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加異丙醇鋁(Al(OiPr)3)的1.0mol/l己烷溶液1.35ml(1.35mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷127g。催化活性為600g/mol·min、所得聚環氧烷的不飽和度為0.005meq/g、分子量分布為1.08、Mh/f為3500g/mol、Mh/3以下的低分子量成分的面積比率為1.4%。
實施例30
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)18g(活性氫量為54mmol)、通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.90g(0.45mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加三苯基鋁(AlPh3)的1.0mol/l二丁醚溶液1.35ml(1.35mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷127g。催化活性為600g/mol·min、所得聚環氧烷的不飽和度為0.006meq/g、分子量分布為1.07、Mh/f為3700g/mol、Mh/3以下的低分子量成分的面積比率為0.4%。
實施例31
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)18g(活性氫量為54mmol)、通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.90g(0.45mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加具有甲基和異丁基的鋁氧烷(Tosoh Finechem Corporation制、MMAO-3A)1.0mol/l己烷溶液1.35ml(1.35mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷85g。催化活性為250g/mol·min、所得聚環氧烷的不飽和度為0.005meq/g、分子量分布為1.05、Mh/f為2000g/mol、Mh/3以下的低分子量成分的面積比率為0.1%。
實施例32
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)18g(活性氫量為54mmol)、通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.90g(0.45mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加二乙基鋅(ZnEt2)的1.0mol/l己烷溶液1.35ml(1.35mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷85g。催化活性為250g/mol·min、所得聚環氧烷的不飽和度為0.005meq/g、分子量分布為1.05、Mh/f為2000g/mol、Mh/3以下的低分子量成分的面積比率為0.1%。
實施例33
將附帶攪拌葉片的0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制SANNIX GP1000)18g(活性氫量為54mmol)、通過合成例2得到的膦腈鹽B的17重量%2-丙醇溶液2.0g(0.45mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加三異丁基鋁(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷127g。催化活性為600g/mol·min、所得聚環氧烷的不飽和度為0.006meq/g、分子量分布為1.06、Mh/f為3700g/mol、Mh/3以下的低分子量成分的面積比率為0.1%。
將上述結果合併示於表8、表9。
[表8]
[表9]
由表8、表9明確得那樣,通過實施例23~例33得到的聚環氧烷滿足下述i)~iv)中的全部條件。
i)不飽和度為0.020meq/g以下
ii)Mw/Mn為1.10以下
iii)Mh/f為1000以上
iv)Mh/3以下的分子量的面積比率為2.0%以下
(其中,將以聚苯乙烯作為標準物質由凝膠滲透色譜測定求出的數均分子量記作Mn,將重均分子量記作Mw、將最高峰的分子量記作Mh,將聚環氧烷的官能團數量記作f。)。
比較例9
將0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)18g(18mmol)、通過合成例1得到的膦腈鹽A的25重量%2-丙醇溶液0.54g(0.27mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷92g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。接著,將高壓釜的內部溫度製成110℃,一邊以反應壓力保持為0.25MPa以下的方式間歇地供給環氧乙烷18g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧乙烷的去除,從而得到無色無臭的聚環氧烷127g。催化活性為500g/mol·min、所得聚環氧烷的不飽和度為0.026meq/g、分子量分布為1.12、Mh/f為3300g/mol、Mh/3以下的低分子量成分的面積比率為3.7%。
比較例10
將0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)18g(18mmol)、三異丁基鋁(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。
將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷10g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。從而得到無色無臭的聚環氧烷18g。催化活性為0g/mol·min、所得聚環氧烷為作為原料的聚醚多元醇(三洋化成工業株式會社制SANNIX GP1000)。
比較例11
將0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)18g(活性氫量為54mmol)、氫氧化鉀(KOH)的50重量%水溶液50mg(0.45mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加三異丁基鋁(TIBAL)的1.0mol/l甲苯溶液1.35ml(1.35mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成90℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷36g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。從而得到無色無臭的聚環氧烷27g。催化活性為60g/mol·min、所得聚環氧烷的不飽和度為0.030meq/g、分子量分布為1.12、Mh/f為500g/mol、Mh/3以下的低分子量成分的面積比率為4.3%。
比較例12
將0.2升高壓釜製成氮氣氣氛,添加聚醚多元醇(三洋化成工業株式會社制、SANNIX GP1000)6g(活性氫量為18mmol)、以及作為膦腈P4鹼的1-叔丁基-4,4,4-三(二甲基氨基)-2,2-雙(三(二甲基氨基)膦烯氨基)-2λ5,4λ5-聯二膦腈的1.0mol/l的己烷溶液18ml(18mmol)。將內部溫度製成80℃,在0.5kPa下進行減壓處理。其後,添加三異丁基鋁(TIBAL)的2.0mol/l甲苯溶液18ml(36mmol),將內部溫度製成80℃,在0.5kPa下進行減壓處理,從而得到環氧烷聚合催化劑。
在所得環氧烷聚合催化劑的存在下,將高壓釜的內部溫度製成20℃,一邊以反應壓力保持為0.3MPa以下的方式間歇地供給環氧丙烷37g,一邊使其反應。反應結束後,在0.5kPa的減壓下進行殘留環氧丙烷的去除。從而得到無色無臭的聚環氧烷42g。催化活性為10g/mol·min、所得聚環氧烷的不飽和度為0.016meq/g、分子量分布為1.36、Mh/f為3,300g/mol、Mh/3以下的低分子量成分的面積比率為3.7%。
將比較例9~12的結果示於表10。
[表10]
應用例1
將通過實施例23得到的聚環氧烷100重量份、二苯基甲烷二異氰酸酯5.8份(商品名:C-1394)、三乙二胺0.05份(商品名:TEDA-L33)投入至デスポカップ中,並進行攪拌。攪拌結束後,使用旋轉型流變儀(UBM公司製造的ソリキッドメーターMR-500),在氮氣氣氛下在25℃、頻率10Hz的條件下測定粘彈性。自攪拌結束起50分鐘後的儲能模量(G』)為45,000Pa。
應用例2
將通過比較例9得到的聚環氧烷100重量份、二苯基甲烷二異氰酸酯5.8份(商品名:C-1394)、三乙二胺0.05份(商品名:TEDA-L33)投入至一次性杯子(diaposable cup)中,並進行攪拌。攪拌結束後,使用旋轉型流變儀(UBM公司製造的ソリキッドメーターMR-500),在氮氣氣氛下在25℃、頻率10Hz的條件下測定粘彈性。自攪拌結束起50分鐘後的儲能模量(G』)為15000Pa。
如上所示,參照特定的實施方式針對本發明進行了詳細說明,但在不超脫本發明本質和範圍的條件下能夠施加各種變更、修正對於本領域技術人員而言是不言而喻的。
本發明中,通式(1)所示的膦腈鹽(Y、R1、R2、X)與路易斯酸與根據需要而使用的含活性氫的化合物的適合組合例如如表11~18所示那樣。
[表11]
[表12]
[表13]
[表14]
[表15]
[表16]
[表17]
[表18]
此外,本發明中,膦腈化合物與路易斯酸與根據需要使用的含活性氫的化合物的適合組合具體而言如表19~24所示那樣。
[表19]
[表20]
[表21]
[表22]
[表23]
[表24]
應予說明,將2014年8月12日申請的日本特願2014-164289號的說明書、權利要求書和摘要、2014年8月12日申請的日本特願2014-164290號的說明書、權利要求書和摘要、2015年7月23日申請的日本特願2015-145645號的說明書、權利要求書和摘要、以及2015年7月23日申請的日本特願2015-145646號的說明書、權利要求書、附圖和摘要的全部內容援引至此,作為本發明說明書的公開內容。
產業上的可利用性
通過使用本發明的環氧烷聚合催化劑,能夠有效地製造聚環氧烷。所得聚環氧烷對於聚氨酯原料、聚酯原料、表面活性劑原料、潤滑劑原料等而言是有用的。
此外,本發明的聚環氧烷對於聚氨酯原料、聚酯原料、表面活性劑原料、潤滑劑原料等而言是有用的。
通過使用本發明的環氧烷聚合催化劑而得到的聚環氧烷尤其是與各種異氰酸酯化合物發生反應,可期待在絕熱材料等中使用的硬質泡沫、汽車的坐墊/椅墊、床上用品等中使用的軟質泡沫、粘接劑、塗料、密封材料、熱固性彈性體、熱塑性彈性體中展開應用。