一種手性的氟烷基取代的哌啶環類化合物的製備方法與流程
2023-09-16 18:51:20 4
本發明涉及有機合成領域,尤其涉及一種手性的氟烷基取代的哌啶環類化合物的製備方法。
背景技術:
不對稱合成研究是手性物質創造的關鍵方法和手段,是化學研究最為活躍的領域之一,它與人類健康的手性醫藥、農藥、香料、香精、食品添加劑以及多種功能材料等領域密切相關,具有重要的理論意義和應用前景。
哌啶環作為一個優勢結構廣泛存在於天然產物和藥物分子中(a.d.elbein,r.molyneux,inalkaloids,chemicalandbiologicalperspectives,(ed.:s.w.pelletier,)johnwiley&sons:newyork,1987,57),現已發現有很多哌啶環特別是手性哌啶環類化合物的藥物,如:喹諾裡西丁類生物鹼((+)-lasubineii)、(s)-哌啶酸、專利s.rault,o.renault,j.guillon,p.dallemagne,p.renard,b.pfeiffer,p.lestage,m.lebrun,e.p.patent1050530,a1公開的記憶增強劑、文獻a.schotte,p.f.m.janssen,w.gommeren,w.h.m.l.luyten,p.vangompel,a.s.lasage,k.deloore,j.e.leysen,psychopharmacology1996,124,57-73中公開的那拉曲坦、專利m.l.childers,p.fuller,d.guerin,j.d.katz,q.pu,m.e.scott,c.f.thompson,m.martinez,d.falcone,l.torres,y.deng,r.kuruklasuriya,h.zeng,y.bai,n.kong,y.liu,z.zheng,w.o.patent2014146491,a1中公開的janus激酶抑制劑,均為手性哌啶環類化合物藥物。而且,在藥物設計與合成中經常在哌啶環上引入含氟基團氟原子或是氟烷基來增加其生物活性。如將這兩者結合起來,發展一種合成氟烷基取代的哌啶環類化合物的方法可能極大拓展藥物範圍,增加藥物活性。最近有報導稱,janus激酶抑制劑的哌啶環的2位被三氟甲基取代後表現出更高的藥物活性。
但是,由於氟烷基取代的底物和合適的手性催化劑的缺乏,如何合成具有手性的氟烷基取代的哌啶環在當前仍是很有挑戰性的課題。
技術實現要素:
有鑑於此,本發明所要解決的技術問題在於提供一種手性的氟烷基取代的哌啶環類化合物的製備方法,本發明提供的製備方法能夠通過一步反應得到含有兩個手性中心的含氟哌啶環類化合物,且產物的立體選擇性高。
本發明提供了一種手性的氟烷基取代的哌啶環類化合物的製備方法,包括:
將式(ii)結構的化合物與式(iii)結構的化合物在式(iv-a)所示催化劑作用下反應,得到式(i-a)結構的化合物;
其中,r1為氫、c1-c30的烷基、c1-c30的烷氧基、滷素、硝基或c4~c30的芳基;
r2為三氟甲基、二氟甲基、單氟甲基或五氟乙基;
r3為c1~c30的烷基或c4~c30的芳香基;
r4為c1~c30的烷基或c4~c15的芳香基;
r5為c1~c30的烷基或c4~c30芳香基;
或者將式(ii)結構的化合物與式(iii)結構的化合物在式(iv-b)所示的催化劑作用下反應,得到式(i-b)結構的化合物;
其中,r1為氫、c1-c30的烷基、c1-c30的烷氧基、滷素、硝基或c4~c30的芳基;
r2為三氟甲基、二氟甲基、單氟甲基或五氟乙基;
r3為c1~c30的烷基或c4~c30的芳香基;
r4為c1~c30的烷基或c4~c15的芳香基;
r5為c1~c30的烷基或c4~c30芳香基。
優選的,所述r1為氫、c3-c15的烷基、c3-c15的烷氧基、滷素、硝基或c6~c15的芳基;
所述r3為c3~c15的烷基或c6~c15的芳香基。
優選的,所述r1為氫、甲基、乙基、丙基、異丙基、丁基、叔丁基、戊基、甲氧基、乙氧基、苯基、萘基、硝基、氟、氯、溴或碘。
優選的,所述r3為甲基、乙基、丙基、異丙基、丁基、叔丁基、羥基甲基、羥基乙基、羥基丙基、羥基丁基、叔丁基二苯基矽醚基乙基、叔丁基二苯基矽醚基丙基、叔丁基二苯基矽醚基丁基、苯基、甲基苯基、甲氧基苯基、2-噻吩基、氰基苯基或氯苯基。
優選的,所述手性的氟烷基取代的哌啶環類化合物為式(i-a-1)、式(i-a-2)、式(i-a-3)、式(i-a-4)、式(i-a-5)、式(i-a-6)、式(i-a-7)、式(i-a-8)、式(i-a-9)、式(i-a-10)、式(i-a-11)、式(i-b-1)、式(i-b-2)、式(i-b-3)、式(i-b-4)、式(i-b-5)、式(i-b-6)、式(i-b-7)、式(i-b-8)、式(i-b-9)、式(i-b-10)或式(i-b-11),
優選的,所述r4為苄基、對氟苄基、對氯苄基、對甲基苄基、對甲氧基苄基、對羥基苄基、苯基、對氟苯基、對氯苯基、對甲基苯基、對甲氧基苯基、對羥基苯基、甲基、乙基、丙基、異丙基、正丁基、仲丁基、異丁基或叔丁基。
優選的,所述r5為苄基、對氟苄基、對氯苄基、對甲基苄基、對甲氧基苄基、對羥基苄基、苯基、對氟苯基、對氯苯基、對甲基苯基、對甲氧基苯基、對羥基苯基、甲基、乙基、丙基、異丙基、正丁基、仲丁基、異丁基、叔丁基、1-羥基-3-苯基丙-2-基、1-羥基-3-對氟苯基丙-2-基、1-羥基-3-對氯苯基丙-2-基、1-羥基-3-對甲基苯基丙-2-基、1-羥基-3-對甲氧基苯基丙-2-基、1-羥基-3-對羥基苯基丙-2-基、1-羥基-2-苯基乙-2-基、1-羥基-2-甲基乙-2-基、1-羥基-2-異丙基乙-2-基或1-羥基-2-叔丁基乙-2-基。
優選的,所述催化劑為式(iv-a-1)、式(iv-a-2)、式(iv-a-3)、式(iv-b-1)、式(iv-b-2)或式(iv-b-3),
優選的,所述式(ii)結構的化合物與所述式(iv-a)所示催化劑的摩爾比為1:(0.05~0.3);
所述式(ii)結構的化合物與所述式(iv-b)所示催化劑的摩爾比為1:(0.05~0.3)。
優選的,所述式(ii)結構的化合物與所述式(iii)結構的化合物的摩爾比為1:(1~5)。
與現有技術相比,本發明提供了一種手性的氟烷基取代的哌啶環類化合物的製備方法,通過將式(ii)結構的化合物與式(iii)結構的化合物在具有式(iv-a)結構或式(iv-b)結構的催化劑作用下反應,得到式(i-a)結構的化合物或式(i-b)結構的化合物,本發明提供的方法通過採用本發明所述的催化劑以及原料使得本發明提供的製備方法通過一步反應製備得到含有兩個手性中心的氟烷基取代的哌啶環類化合物,製備方法簡單,且得到的產物的立體選擇性高,產率高,實驗結果表明,本發明提供的製備方法中,產品的收率在90%以上,ee值在89%以上。
附圖說明
圖1為本發明實施例1製備得到的產物的xrd單晶結構圖;
圖2為本發明實施例1提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖3為本發明實施例1提供的加成產物的13c核磁共振(13cnmr)譜圖;
圖4為本發明實施例2提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖5為本發明實施例2提供的加成產物的13c核磁共振(13cnmr)譜圖;
圖6為本發明實施例3提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖7為本發明實施例3提供的加成產物的13c核磁共振(13cnmr)譜圖;
圖8為本發明實施例4提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖9為本發明實施例4提供的加成產物的13c核磁共振(13cnmr)譜圖;
圖10為本發明實施例5提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖11為本發明實施例5提供的加成產物的13c核磁共振(13cnmr)譜圖;
圖12為本發明實施例6提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖13為本發明實施例6提供的加成產物的13c核磁共振(13cnmr)譜圖;
圖14為本發明實施例7提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖15為本發明實施例7提供的加成產物的13c核磁共振(13cnmr)譜圖;
圖16為本發明實施例8提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖17為本發明實施例8提供的加成產物的13c核磁共振(13cnmr)譜圖;
圖18為本發明實施例9提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖19為本發明實施例9提供的加成產物的13c核磁共振(13cnmr)譜圖;
圖20為本發明實施例10提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖21為本發明實施例10提供的加成產物的13c核磁共振(13cnmr)譜圖;
圖22為本發明實施例11提供的加成產物的1h核磁共振(1hnmr)譜圖;
圖23為本發明實施例11提供的加成產物的13c核磁共振(13cnmr)譜圖。
具體實施方式
本發明還提供了一種本發明所述的手性的氟烷基取代的哌啶環類化合物的製備方法,包括:
將式(ii)結構的化合物與式(iii)結構的化合物在式(iv-a)所示催化劑作用下反應,得到式(i-a)結構的化合物;
其中,r1為氫、c1-c30的烷基、c1-c30的烷氧基、滷素、硝基或c4~c30的芳基;
r2為三氟甲基、二氟甲基、單氟甲基或五氟乙基;
r3為c1~c30的烷基或c4~c30的芳香基;
r4為c1~c30的烷基或c4~c15的芳香基;
r5為c1~c30的烷基或c4~c30芳香基;
或者將式(ii)結構的化合物與式(iii)結構的化合物在式(iv-b)所示的催化劑作用下反應,得到式(i-b)結構的化合物;
其中,r1為氫、c1-c30的烷基、c1-c30的烷氧基、滷素、硝基或c4~c30的芳基;
r2為三氟甲基、二氟甲基、單氟甲基或五氟乙基;
r3為c1~c30的烷基或c4~c30的芳香基;
r4為c1~c30的烷基或c4~c15的芳香基;
r5為c1~c30的烷基或c4~c30芳香基。
按照本發明,本發明將式(ii)結構的化合物與式(iii)結構的化合物在式(iv-a)所示催化劑作用下反應,得到式(i-a)結構的化合物;或者將式(ii)結構的化合物與式(iii)結構的化合物在式(iv-b)所示的催化劑作用下反應,得到式(i-b)結構的化合物;其中,所述式(ii)結構的化合物與所述式(iii)結構的化合物的摩爾比優選為1:(1~5),更優選為1:(2~4),最優選為1:(2.5~3);所述式(ii)結構的化合物與所述式(iv-a)所示催化劑的摩爾比為1:(0.05~0.3),更優選為1:(0.1~0.2);所述式(ii)結構的化合物與所述式(iv-b)所示催化劑的摩爾比為1:(0.05~0.3),更優選為1:(0.1~0.2);所述反應的溶劑優選為甲苯、二甲苯、氯仿、二氯甲烷、乙酸乙酯、四氫呋喃、乙醚、乙腈、甲醇、乙醇和異丙醇中的一種或幾種,更優選為甲苯、二甲苯、氯仿、二氯甲烷、乙酸乙酯、四氫呋喃、乙醚或乙腈;所述反應的溫度優選為0~50℃,更優選為10~40℃,最優選為20~30℃;且為了使反應能夠更順利的進行,本發明優選還加入酸性添加劑,所述酸性添加劑優選為對硝基苯甲酸、乙酸、特戊酸、三氟乙酸、苯甲酸、苯乙酸、對甲氧基苯甲酸、對甲基苯甲酸、對氯苯甲酸和對氟苯甲酸中的一種或是幾種,所述式(ii)結構的化合物與所述酸性添加劑的摩爾比為1:(0.05~0.3),更優選為1:(0.1~0.2)。
本發明中,所述式(ii)結構的化合物中,所述r1優選為氫、c3-c15的烷基、c3-c15的烷氧基、滷素、硝基或c6~c15的芳基,更優選為氫、甲基、乙基、丙基、異丙基、丁基、叔丁基、戊基、甲氧基、乙氧基、苯基、萘基、硝基、氟、氯、溴或碘,具體的,所述化合物為式(ii-1)、式(ii-2)、式(ii-3)、式(ii-4)、式(ii-5)、式(ii-6)、式(ii-7)、式(ii-8)、式(ii-9)、式(ii-10)或式(ii-11),
本發明中,所述式(iii)結構的化合物中,所述r3優選為c3~c15的烷基或c6~c15的芳香基,更優選為甲基、乙基、丙基、異丙基、丁基、叔丁基、羥基甲基、羥基乙基、羥基丙基、羥基丁基、叔丁基二苯基矽醚基乙基、叔丁基二苯基矽醚基丙基、叔丁基二苯基矽醚基丁基、苯基、甲基苯基、甲氧基苯基、2-噻吩基、氰基苯基或氯苯基。
更具體的,所述手性的氟烷基取代的哌啶環類化合物為式(i-a-1)、式(i-a-2)、式(i-a-3)、式(i-a-4)、式(i-a-5)、式(i-a-6)、式(i-a-7)、式(i-a-8)、式(i-a-9)、式(i-a-10)、式(i-a-11)、式(i-b-1)、式(i-b-2)、式(i-b-3)、式(i-b-4)、式(i-b-5)、式(i-b-6)、式(i-b-7)、式(i-b-8)、式(i-b-9)、式(i-b-10)或式(i-b-11),
本發明所述的催化劑中,所述r4優選為c3~c15的烷基或c4~c15的芳香基,更優選為苄基、對氟苄基、對氯苄基、對甲基苄基、對甲氧基苄基、對羥基苄基、苯基、對氟苯基、對氯苯基、對甲基苯基、對甲氧基苯基、對羥基苯基、甲基、乙基、丙基、異丙基、正丁基、仲丁基、異丁基或叔丁基;所述r5優選為c3~c15的烷基或c3~c15芳香基,更優選為苄基、對氟苄基、對氯苄基、對甲基苄基、對甲氧基苄基、對羥基苄基、苯基、對氟苯基、對氯苯基、對甲基苯基、對甲氧基苯基、對羥基苯基、甲基、乙基、丙基、異丙基、正丁基、仲丁基、異丁基、叔丁基、1-羥基-3-苯基丙-2-基、1-羥基-3-對氟苯基丙-2-基、1-羥基-3-對氯苯基丙-2-基、1-羥基-3-對甲基苯基丙-2-基、1-羥基-3-對甲氧基苯基丙-2-基、1-羥基-3-對羥基苯基丙-2-基、1-羥基-2-苯基乙-2-基、1-羥基-2-甲基乙-2-基、1-羥基-2-異丙基乙-2-基或1-羥基-2-叔丁基乙-2-基,更具體的,所述催化劑具有式(iv-a-1)、式(iv-a-2)、式(iv-a-3)、式(iv-b-1)、式(iv-b-2)或式(iv-b-3)結構,
按照本發明,本發明對得到的反應產物的分離純化方法沒有特殊要求,可以為柱層色譜、液相色譜、蒸餾或重結晶更優選地,所述分離提純方式為柱層色譜,所述洗脫劑優選為石油醚和乙醚。
本發明提供的一種手性的氟烷基取代的哌啶環類化合物的製備方法,通過採用本發明所述的催化劑以及原料使得本發明提供的製備方法通過一步反應製備得到含有兩個手性中心的氟烷基取代的哌啶環類化合物,製備方法簡單,且得到的產物的立體選擇性高,產率高。而且,本發明提供的一種手性的氟烷基取代的哌啶環類化合物,具有式(i-a)結構或式(i-b)結構,本發明提供的化合物具有兩個手性中心,而且化合物中的異噻唑環可以發生開環反應,且哌啶環上還有羰基官能團,所以,該化合物可以衍生出很多重要的藥物中間體或者藥物,是具有廣闊應用前景的原料。
下面將結合本發明實施例的技術方案進行清楚、完整地描述,顯然,所描述的實施例僅僅是本發明一部分實施例,而不是全部的實施例。基於本發明中的實施例,本領域普通技術人員在沒有做出創造性勞動前提下所獲得的所有其他實施例,都屬於本發明保護的範圍。
在實施例中所使用的催化劑、氟烷基取代的縮醛胺及烯酮為根據文獻方法合成(a)s.zhang,l.li,y.hu,y.li,y.yu,z.zha,z.wang,org.lett.2015,17,5036-5039.b)y.liu,t.kang,q.liu,l.chen,y.wang,j.liu,y.xie,j.yang,l.he,org.lett.2013,15,6090-6093.),酸性添加劑等購於韶遠韶遠化學科技(上海)有限公司。所有試劑均為直接購買的分析純試劑,使用前未經其他處理,所用溶劑或洗脫劑購於國藥。
實施例1:
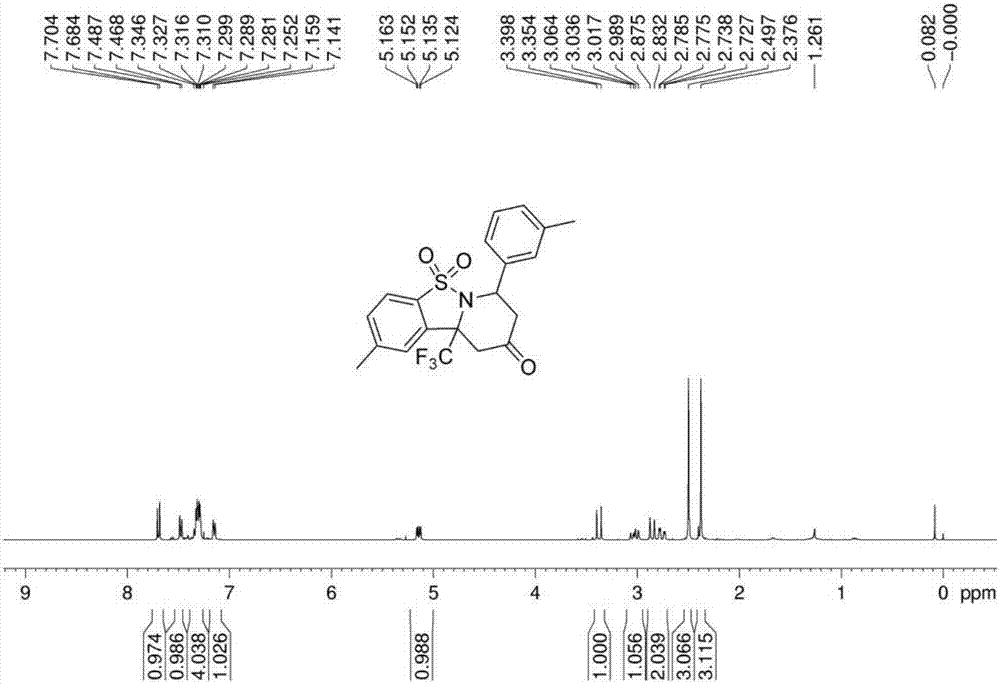
在一個20ml反應管中,加入式式(iv-a-3)所示催化劑(2s,3s)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-甲基苯丙醯胺(0.06mmol,15.9mg)、對硝基苯甲酸(0.06mmol,10.0mg),式(ii-2)所示氟烷基取代的縮醛胺3-(三氟甲基)3-羥基-5-甲基2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,80.2mg)、烯酮(e)-4-(3-甲基苯基)-丁-3-烯-2-酮(0.6mmol,96.1mg)和氯仿(3.0ml),在30℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物(7r,10ar)-2-甲基-7-(3-甲基苯基)-10a-(三氟甲基)-10,10a-二氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-9(8h)-酮5,5-二氧,產率為99%。
通過xrd單晶衍射對產物的絕對構型進行了測定,其結果參見圖1,圖1為本發明實施例1製備得到的產物的xrd單晶結構圖,其數據見表1,表1為本發明實施例1製備的產物的xrd單晶結構的相關數據,
表1為本發明實施例1製備的產物的xrd單晶結構的相關數據
收集/獨立衍射數據:7031/3437(rint=0.0195),觀測次數[i>2σ(i)]3305,參數255.可觀察衍射的吻合因子[i>2σ(i)]:r1=0.0351,wr2=0.0927;所有數據的吻合因子:r1=0.0338,wr2=0.0904;flack參數=-0.017(10)。
用核磁共振波譜儀對所述加成產物進行分析,結果參見圖2~3,圖2為本發明實施例1提供的加成產物的1h核磁共振(1hnmr)譜圖;圖3為本發明實施例1提供的加成產物的13c核磁共振(13cnmr)譜圖,從圖中可知,其表徵數據為:
1hnmr(400mhz,cdcl3):δ7.70-7.68(d,j=8.0hz,1h),7.49-7.47(d,j=8.0hz,1h),7.35-7.28(m,4h),7.16-7.14(d,j=7.1hz,1h),5.16-5.12(dd,j=4.6hz,11.3hz,1h),3.40-3.35(d,j=17.2hz,1h),3.06-2.99(dd,j=11.4hz,19.0hz,1h),2.88-2.83(d,j=17.2hz,1h),2.79-2.73(dd,j=4.0hz,18.9hz,1h),2.50(s,3h),2.08(s,3h);
13cnmr(100mhz,cdcl3):δ200.93,145.31,138.97,138.44,133.21,132.71,132.10,129.32,128.74,128.20-122.76(q,j=284.1hz),127.67,124.44-124.43(d,j=1.4hz),124.02,121.50,67.71-66.79(q,j=30.6hz),56.72,45.52-45.50(d,j=2.2hz),43.43,21.83,21.43;
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):2923,1735,1600,1490,1324,1205,1178,1154,1066,953,786,706,683;
通過高分辨質譜對得到的產物進行檢測,結果表明,其hrms(esi)m/z理論值為c20h18f3no3s[m+na]+432.0857,實際值為432.0860。
通過上述檢測結果可知,本發明實施例1提供的加成產物的結構為式(i-a-1)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為90%;
通過1hnmr測得所述加成產物的dr值為11:1。
實施例2:
在一個20ml反應管中,加入式(iv-a-1)所示催化劑(s)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-苯丙醯胺(0.06mmol,17.9mg)、對硝基苯甲酸(0.06mmol,10.0mg),式(ii-1)所示氟烷基取代的縮醛胺3-(三氟甲基)3-羥基-2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,76.0mg)、烯酮(e)-4-苯基-丁-3-烯-2-酮(0.6mmol,87.7mg)和氯仿(3.0ml),在30℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物(7r,10ar)-7-苯基-10a-(三氟甲基)-10,10a-二氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-9(8h)-酮5,5-二氧,產率為90%。
通過用核磁共振波譜儀對所述加成產物進行分析,結果參見圖4~5,圖4為本發明實施例2提供的加成產物的1h核磁共振(1hnmr)譜圖;圖5為本發明實施例2提供的加成產物的13c核磁共振(13cnmr)譜圖,由圖可知,其表徵數據為:1hnmr(400mhz,d6-acetone):δ7.92-7.91(m,3h),7.89-7.83(m,1h),7.57-7.55(d,j=7.3hz),7.43-7.40(m,2h),7.36-7.32(m,1h),5.36-5.32(dd,j=5.0hz,11.4hz,1h),3.63-3.58(d,j=17.2hz,1h),3.45-3.41(dd,j=0.8hz,17.2hz,1h),2.95-2.79(m,2h);
13cnmr(100mhz,d6-acetone):δ201.57,141.41,135.94,135.09,133.94,132.98,129.96-124.29(q,j=283.6hz),129.44,128.98,128.11,126.01-125.99(d,j=1.8hz),122.20,69.12-68.52(q,j=30.0hz),58.18,47.02-46.99(d,j=2.7hz),43.90;
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):2924,1733,1455,1410,1327,1222,1182,1065,990,947,763,702,668;
通過高分辨質譜對得到的產物進行檢測,結果表明,其hrms(esi)m/z理論值為c18h14f3no3s[m+na]+404.0544,實際值為404.0545。
通過上述檢測結果可知,本發明實施例2提供的加成產物的結構為式(i-a-2)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為92%;
通過1hnmr測得所述加成產物的dr值為27:1。
實施例3:
在一個20ml反應管中,加入式(iv-a-3)所示催化劑(2s,3s)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-甲基苯丙醯胺(0.06mmol,15.9mg)、對硝基苯甲酸(0.06mmol,10.0mg),式(ii-2)所示氟烷基取代的縮醛胺3-(三氟甲基)3-羥基-5-甲基-2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,80.2mg)、烯酮(e)-4-(3,4-二甲氧基苯基)-丁-3-烯-2-酮(0.6mmol,123.7mg)和氯仿(3.0ml),在30℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物(7r,10ar)-2-甲基-7-(3,4-二甲氧基苯基)-10a-(三氟甲基)-10,10a-二氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-9(8h)-酮5,5-二氧,產率為99%。
通過用核磁共振波譜儀對所述加成產物進行分析,結果參見圖6~7,圖6為本發明實施例3提供的加成產物的1h核磁共振(1hnmr)譜圖;圖7為本發明實施例3提供的加成產物的13c核磁共振(13cnmr)譜圖,由圖可知,其表徵數據為:1hnmr(400mhz,cdcl3):δ7.73-7.71(d,j=8.0hz,1h),7.50-7.48(d,j=8.0hz,1h),7.33(s,1h),7.09-7.08(d,j=2.0hz,1h),7.05-7.02(dd,j=2.1hz,8.3hz,1h),6.90-6.88(d,j=8.3hz,1h),5.21-5.17(dd,j=4.8hz,10.9hz,1h),3.89(s,3h),3.88(s,3h),3.40-3.36(d,j=17.2hz,1h),3.08-3.00(dd,j=11.0hz,18.7hz,1h),2.87-2.83(d,j=17.3hz,1h),2.82-2.76(dd,j=5.0hz,19.1hz,1h),2.51(s,3h);
13cnmr(100mhz,cdcl3):δ200.91,149.03,148.93,145.31,133.09,132.67,131.90,131.37,125.55-122.71(q,j=284.3hz),124.41,121.41,119.07,110.98,110.16,67.28-66.66(q,j=30.6hz),56.17,55.69,45.39,43.39,21.77;
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):2936,1733,1597,1518,1466,1422,1324,1266,1205,1177,1155,1065,1026,954,815,766,739,709,684;
通過高分辨質譜對得到的產物進行檢測,結果表明,其hrms(esi)m/z理論值為c21h20f3no5s[m+na]+478.0912,實際值478.0917。
通過上述檢測結果可知,本發明實施例3提供的加成產物的結構為式(i-a-3)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為92%。
通過1hnmr測得所述加成產物的dr值為11:1。
實施例4:
在一個20ml反應管中,加入式(iv-a-3)所示催化劑(2s,3s)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-甲基苯丙醯胺(0.06mmol,15.9mg)、對硝基苯甲酸(0.06mmol,10.0mg),式(ii-2)所示氟烷基取代的縮醛胺3-(三氟甲基)3-羥基-5-甲基-2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,80.2mg)、烯酮(e)-4-(噻吩-2-基)-丁-3-烯-2-酮(0.6mmol,91.3mg)和氯仿(3.0ml),在30℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物(7r,10ar)-2-甲基-7-(噻吩-2-基)-10a-(三氟甲基)-10,10a-二氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-9(8h)-酮5,5-二氧,產率為91%。
通過用核磁共振波譜儀對所述加成產物進行分析,結果參見圖8~9,圖8為本發明實施例4提供的加成產物的1h核磁共振(1hnmr)譜圖;圖9為本發明實施例4提供的加成產物的13c核磁共振(13cnmr)譜圖,由圖可知。其表徵數據為:1hnmr(400mhz,cdcl3):δ7.76-7.74(d,j=8.0hz,1h),7.52-7.50(m,1h),7.37-7.36(m,1h),7.31(s,1h),7.22-7.21(m,1h),7.02-6.99(m,1h),5.66-5.62(dd,j=5.3hz,9.2hz,1h),3.41-3.37(d,j=17.1hz,1h),3.21-3.14(dd,j=9.3hz,18.6hz,1h),2.95-2.89(dd,j=5.2hz,18.3hz,1h),2.86-2.82(d,j=17.1hz,1h),2.52(s,3h);
13cnmr(100mhz,cdcl3):δ200.18,145.53,142.44,133.15,132.79,132.01,126.86,126.79,126.49,125.43-122.59(q,j=284.0hz),124.54,121.68,67.83-66.90(q,j=31.0hz),51.82,45.22,43.32,21.94;
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):2927,1733,1598,1321,1273,1205,1170,1154,1064,950,822,710,666;
通過高分辨質譜對得到的產物進行檢測,結果表明,其hrms(esi)m/z理論值為c17h14f3no3s2[m+na]+424.0265,實際值為424.0263。
通過上述檢測結果可知,本發明實施例4提供的加成產物的結構為式(i-a-4)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為89%。
通過1hnmr測得所述加成產物的dr值為22:1。
實施例5:
在一個20ml反應管中,加入式(iv-a-3)所示催化劑(2s,3s)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-甲基苯丙醯胺(0.03mmol,8.0mg)、對硝基苯甲酸(0.03mmol,5.0mg),式(ii-6)所示氟烷基取代的縮醛胺3-(三氟甲基)3-羥基-5-氯-2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,86.3mg)、烯酮(e)-4-苯基-丁-3-烯-2-酮(0.6mmol,87.7mg)和氯仿(3.0ml),在30℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物(7r,10ar)-2-氯-7-苯基-10a-(三氟甲基)-10,10a-二氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-9(8h)-酮5,5-二氧,產率為86%。
通過用核磁共振波譜儀對所述加成產物進行分析,結果參見圖10~11,圖10為本發明實施例5提供的加成產物的1h核磁共振(1hnmr)譜圖;圖11為本發明實施例5提供的加成產物的13c核磁共振(13cnmr)譜圖,由圖可知。其表徵數據為:1hnmr(400mhz,d6-acetone):δ8.05(s,1h),7.97-7.95(d,j=8.3hz,1h),7.90-7.87(m,1h),7.56-7.54(m,2h),7.44-7.40(m,2h),7.37-7.33(m,1h),5.37-5.33(dd,j=5.1hz,11.2hz,1h),3.72-3.68(d,j=17.2hz,1h),3.53-3.49(d,j=17.3hz,1h),2.95-2.80(m,2h);
13cnmr(100mhz,d6-acetone):δ201.27,141.21,140.77,136.08,134.81,133.48,129.77-124.10(q,j=283.6hz),129.52,129.10,128.14,126.36-126.35(d,j=1.6hz),123.97,68.65-68.35(q,j=30.4hz),58.45,47.08-47.05(d,j=2.6hz),43.57;
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):1735,1583,1459,1406,1328,1222,1182,1094,1062,999,952,898,830,763,702,673;
通過高分辨質譜對得到的產物進行檢測,結果表明,其hrms(esi)m/z理論值c18h13clf3no3s[m+na]+438.0154,實際值為438.0152。
通過上述檢測結果可知,本發明實施例5提供的加成產物的結構為式(i-a-5)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為97%。
通過1hnmr測得所述加成產物的dr值為46:1。
實施例6:
在一個20ml反應管中,加入式(iv-a-3)所示催化劑(2s,3s)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-甲基苯丙醯胺(0.06mmol,15.9mg)、2,6-二氟苯甲酸(0.06mmol,9.5mg),式(ii-7)所示氟烷基取代的縮醛胺3-(三氟甲基)3-羥基-4-氟-2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,81.4mg)、烯酮(e)-4-苯基-丁-3-烯-2-酮(0.6mmol,87.7mg)和氯仿(3.0ml),在30℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物(7r,10ar)-1-氟-7-苯基-10a-(三氟甲基)-10,10a-二氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-9(8h)-酮5,5-二氧,產率為96%。
通過用核磁共振波譜儀對所述加成產物進行分析,結果參見圖12~13,圖12為本發明實施例6提供的加成產物的1h核磁共振(1hnmr)譜圖;圖13為本發明實施例6提供的加成產物的13c核磁共振(13cnmr)譜圖,由圖可知,其表徵數據為:1hnmr(400mhz,d6-acetone):δ7.95-7.89(m,1h),7.79-7.77(d,j=7.6hz,1h),7.71-7.66(m,1h),7.56-7.54(d,j=7.4hz,2h),7.44-7.40(dd,j=7.1hz,hz,2h),7.36-7.33(m,1h),5.35-5.31(dd,j=4.9hz,11.4hz,1h),3.69-3.59(m,2h),2.95-2.80(m,2h);
13cnmr(100mhz,d6-acetone):δ201.02,159.81-157.26(d,j=255.4hz),140.99,138.77-138.75(d,j=2.0hz),136.14-136.06(d,j=8.0hz),129.77-124.08(q,j=282.3hz),129.45,129.08,128.08,122.56-122.34(d,j=21.8hz),120.51-120.35(d,j=16.8hz),118.65-118.61(d,j=4.0hz),68.62-67.67(m),58.48,47.06-47.04(d,j=2.3hz),42.04-42.00(d,j=3.6hz);
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):2924,1735,1608,1473,1412,1331,1259,1202,1104,1003,989,951,913,797,762,725,700,666;
通過高分辨質譜對得到的產物進行檢測,結果表明,hrms(esi)m/z理論值c18h13f4no3s[m+na]+422.0450,實際值422.0454。
通過上述檢測結果可知,本發明實施例6提供的加成產物的結構為式(i-a-6)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為90%。
通過1hnmr測得所述加成產物的dr值為75:1。
實施例7:
在一個20ml反應管中,加入式(iv-a-3)所示催化劑(2s,3s)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-甲基苯丙醯胺(0.06mmol,15.9mg)、對硝基苯甲酸(0.06mmol,10.0mg),式(ii-9)所示氟烷基取代的縮醛胺3-(二氟甲基)3-羥基-5-甲基-2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,74.8mg)、烯酮(e)-4-苯基-丁-3-烯-2-酮(0.6mmol,87.7mg)和氯仿(3.0ml),在30℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物(7r,10ar)-7-苯基-10a-(二氟甲基)-10,10a-二氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-9(8h)-酮5,5-二氧,產率為95%。
通過用核磁共振波譜儀對所述加成產物進行分析,結果參見圖14~15,圖14為本發明實施例7提供的加成產物的1h核磁共振(1hnmr)譜圖;圖15為本發明實施例7提供的加成產物的13c核磁共振(13cnmr)譜圖,由圖可知,其表徵數據為:1hnmr(400mhz,cdcl3):δ7.74-7.72(d,j=8.0hz,1h),7.54-7.53(d,j=7.5hz,2h),7.48-7.46(d,j=8.0hz,1h),7.44-7.40(t,j=7.2hz,2h),7.37-7.35(m,1h),7.29(s,1h),5.78-5.50(t,jf-h=55.4hz,1h),5.37-5.34(t,j=6.7hz,1h),3.27-3.23(d,j=16.8hz,1h),3.16-3.09(dd,j=8.2hz,17.9hz,1h),2.85-2.76(q,2h),2.50(s,3h);
13cnmr(100mhz,cdcl3):δ202.16,145.23,138.95,134.77-134.75(d,j=3.0hz),132.23,132.05,128.95,128.51,127.17,124.98,121.41,117.02-111.99(t,j=251.5hz),67.12-66.64(t,j=23.5hz),55.25,44.03-44.00(d,j=3.0hz),42.32,21.91;
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):1730,1599,1494,1456,1372,1311,1225,1193,1166,1076,998,919,818,761,688;
通過高分辨質譜對得到的產物進行檢測,結果表明,hrms(esi)m/z理論值c19h17f2no3s[m+na]+400.0795,實際值為400.0799。
通過上述檢測結果可知,本發明實施例7提供的加成產物的結構為式(i-a-7)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為94%。
通過1hnmr測得所述加成產物的dr值為4:1。
實施例8:
在一個20ml反應管中,加入式(iv-a-3)所示催化劑(2s,3s)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-甲基苯丙醯胺(0.06mmol,15.9mg)、對硝基苯甲酸(0.06mmol,10.0mg),式(ii-10)所示氟烷基取代的縮醛胺3-五氟乙基)3-羥基-5-甲基-2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,95.2mg)、烯酮(e)-4-苯基-丁-3-烯-2-酮(0.6mmol,87.7mg)和氯仿(3.0ml),在30℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物(7r,10ar)-7-苯基-10a-(五氟乙基)-10,10a-二氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-9(8h)-酮5,5-二氧,產率為99%。
通過用核磁共振波譜儀對所述加成產物進行分析,結果參見圖16~17,圖16為本發明實施例8提供的加成產物的1h核磁共振(1hnmr)譜圖;圖17為本發明實施例8提供的加成產物的13c核磁共振(13cnmr)譜圖,由圖可知,其表徵數據為:1hnmr(400mhz,d6-acetone):δ7.79-7.77(d,j=8.0hz,1h),7.74-7.73(d,j=4.1hz,1h),7.67-7.65(m,1h),7.56-7.54(m,2h),7.42-7.38(m,2h),7.35-7.31(m,1h),5.36-5.32(dd,j=4.4hz,12.4hz,1h),3.66-3.61(dd,j=1.8hz,17.1hz,1h),3.34-3.29(d,j=17.2hz),2.97-2.88(m,1h),2.81-2.74(m,1h),2.54(s,3h);
13cnmr(100mhz,d6-acetone):δ201.66,146.52,141.39,134.34-134.29(d,j=5.2hz),133.84,132.77,129.45,129.01,128.37,126.49-126.43(d,j=6.7hz),122.08,121.78-112.96(m),69.19-68.69(q,j=23.1hz),58.84,46.88-46.81(d,j=6.9hz),44.91,21.79;
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):2927,1735,1599,1328,1227,1167,1144,1019,932,819,761;
通過高分辨質譜對得到的產物進行檢測,結果表明,hrms(esi)m/z理論值c20h16f5no3s[m+na]+468.0669,實際值為468.0673。
通過上述檢測結果可知,本發明實施例8提供的加成產物的結構為式(i-a-8)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為98%;
通過1hnmr測得所述加成產物的dr值為3:1。
實施例9:
在一個20ml反應管中,加入式(iv-a-3)所示催化劑(2s,3s)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-甲基苯丙醯胺(0.06mmol,15.9mg)、對硝基苯甲酸(0.06mmol,10.0mg),式(ii-2)所示氟烷基取代的縮醛胺3-(三氟甲基)3-羥基-5-甲基-2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,80.2mg)、烯酮(e)-7-((叔丁基-二苯基矽基)氧)庚-3-烯-2-酮(0.9mmol,329.9mg)和氯仿(4.0ml),在20℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物(7r,10ar)-2-甲基-7-(3-((叔丁基-二苯基矽基)氧)丙基)苯基-10a-(三氟甲基)-10,10a-二氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-9(8h)-酮5,5-二氧,產率為93%。
通過用核磁共振波譜儀對所述加成產物進行分析,結果參見圖18~19,圖18為本發明實施例9提供的加成產物的1h核磁共振(1hnmr)譜圖;圖19為本發明實施例9提供的加成產物的13c核磁共振(13cnmr)譜圖,由圖可知,其表徵數據為:1hnmr(400mhz,cdcl3):δ7.69-7.67(m,5h),7.47-7.37(m,7h),7.32(s,1h),4.06-4.02(m,1h),3.80-3.77(t,j=6.1hz,2h),3.23-3.19(d,j=15.4hz,1h),2.93-2.82(m,2h),2.65-2.61(m,1h),2.51-2.46(m,4h),2.22-2.16(m,1h),1.91-1.80(m,2h),1.07(s,9h);
13cnmr(100mhz,cdcl3):δ202.17,145.29,135.53,133.76,133.67,132.96,132.56,132.21,129.60,128.91-123.21(q,j=285.0hz),127.65,124.39-124.37(d,j=1.6hz),121.23,67.56-66.65(q,j=30.4hz),62.86,55.83,44.10,29.64,29.49,27.73,26.84,21.91,19.16;
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):2931,2858,1730,1427,1315,1257,1172,1108,1006,821,739,704,683;
通過高分辨質譜對得到的產物進行檢測,結果表明,hrms(esi)m/z理論值c32h36f3no4ssi[m+na]+638.1984,實際值為638.1988。
通過上述檢測結果可知,本發明實施例9提供的加成產物的結構為式(i-a-9)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為93%;
通過1hnmr測得所述加成產物的dr值為9:1。
實施例10:
在一個20ml反應管中,加入式(iv-a-3)所示催化劑(2s,3s)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-甲基苯丙醯胺(0.06mmol,15.9mg)、對硝基苯甲酸(0.06mmol,10.0mg),式(ii-8)所示氟烷基取代的縮醛胺3-(三氟甲基)3-羥基-6-甲基-2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,80.2mg)、烯酮(e)-4-(4-氰基苯基)-丁-3-烯-2-酮(0.6mmol,102.7mg)和氯仿(3.0ml),在30℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物4-(7r,10ar)-3-甲基-5,5-二氧-9-氧-10a-(三氟甲基)-8,9,10,10a-四氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-7-基)苯甲腈,產率為97%。
通過用核磁共振波譜儀對所述加成產物進行分析,結果參見圖20~21,圖20為本發明實施例10提供的加成產物的1h核磁共振(1hnmr)譜圖;圖21為本發明實施例10提供的加成產物的13c核磁共振(13cnmr)譜圖,由圖可知,其表徵數據為:1hnmr(400mhz,cdcl3):δ7.76-7.73(m,2h),7.67-7.65(d,j=8.4hz,2h),7.64(s,1h),7.59-7.57(m,1h),7.45-7.43(d,j=8.2hz,1h),5.24-5.20(dd,j=4.6hz,11.6hz,1h),3.45-3.41(d,j=17.3hz,1h),2.99-2.96(m,1h),2.87-2.82(m,2h),2.51(s,3h);
13cnmr(100mhz,cdcl3):δ199.50,144.18,143.22,135.47,134.12,132.87,129.94,127.92,125.57-122.73(q,j=284.1hz),124.06,121.83,118.35,112.69,67.39-66.77(q,j=30.8hz),56.37,44.89,43.66,21.36;
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):2925,1733,1610,1499,1414,1325,1226,1159,1122,1065,990,948,843,735,706,670;
通過高分辨質譜對得到的產物進行檢測,結果表明,hrms(esi)m/z理論值c20h15f3n2o3s[m+na]+443.0653,實際值為443.0648。
通過上述檢測結果可知,本發明實施例10提供的加成產物的結構為式(i-a-10)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為94%.
通過1hnmr測得所述加成產物的dr值為10:1。
實施例11:
在一個20ml反應管中,加入式(iv-b-1)所示催化劑(r)-2-氨基-n-((s)-1-苯基-3-羥基丙-2-基)-3-苯丙醯胺(0.06mmol,17.9mg)、對硝基苯甲酸(0.06mmol,10.0mg),式(ii-2)所示氟烷基取代的縮醛胺3-(三氟甲基)3-羥基-5-甲基2,3-二氫苯丙[d]異噻唑1,1-二氧(0.3mmol,80.2mg)、烯酮(e)-4-苯基-丁-3-烯-2-酮(0.6mmol,87.7mg)和氯仿(3.0ml),在40℃攪拌反應。反應完成後(tlc跟蹤檢測),旋幹得到的殘留物用乙酸乙酯/石油醚體系作為洗脫劑過色譜柱得到產物(7s,10as)-2-甲基-7-苯基-10a-(三氟甲基)-10,10a-二氫-7h-苯並[4,5]異噻唑並[2,3-a]吡啶-9(8h)-酮5,5-二氧,產率為99%。
通過用核磁共振波譜儀對所述加成產物進行分析,結果參見圖22~23,圖22為本發明實施例11提供的加成產物的1h核磁共振(1hnmr)譜圖;圖23為本發明實施例11提供的加成產物的13c核磁共振(13cnmr)譜圖,由圖可知,其表徵數據為:1hnmr(400mhz,cdcl3):δ7.72-7.70(d,j=8.0hz,1h),7.53-7.48(m,3h),7.44-7.40(m,2h),7.37-7.31(m,2h),5.21-5.17(dd,j=4.5hz,11.1hz,1h),3.41-3.37(d,j=17.2hz,1h),3.09-3.02(dd,j=11.3hz,18.8hz,1h),2.88-2.84(d,j=17.2hz,1h),2.82-2.76(dd,j=4.1hz,19.0hz,1h),2.51(s,3h);
13cnmr(100mhz,cdcl3):δ200.83,145.38,139.00,133.26,132.76,132.08,128.89,128.61,127.10,125.63-122.79(q,j=284.1hz),124.47,121.59,67.74-66.82(q,j=30.8hz),56.72,45.46,43.53,21.89;
通過紅外光譜對得到的產物進行檢測,其結果為:ir(film,ν/cm-1):2924,1735,1704,1688,1656,1599,1561,1544,1510,1459,1399,1366,1323,1260,1222,1205,1177,1104,1063,995,953,819,767,700,683;
通過高分辨質譜對得到的產物進行檢測,結果表明,其hrms(esi)m/z理論值為c19h16f3no3s[m+na]+418.0701,實際值為418.0705。
通過上述檢測結果可知,本發明實施例11提供的加成產物的結構為式(i-b-11)所示的結構。
對所述加成產物通過用hplc進行測定,測得其ee值為89%;
通過1hnmr測得所述加成產物的dr值為10:1。
以上實施例的說明只是用於幫助理解本發明的方法及其核心思想。應當指出,對於本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以對本發明進行若干改進和修飾,這些改進和修飾也落入本發明權利要求的保護範圍內。