鋯的吡啶絡合物、包含所述鋯的吡啶絡合物的催化體系以及使共軛二烯(共)聚合的工藝的製作方法
2023-05-20 11:42:41 8
本發明涉及鋯的吡啶絡合物(pyridine complex of zirconium)。
更特別地,本發明涉及鋯的吡啶絡合物並且涉及其在用於使共軛二烯(conjugated diene)(共)聚合的催化體系中的用途。
本發明還涉及包含所述鋯的吡啶絡合物的用於使共軛二烯(共)聚合的催化體系。
另外,本發明涉及用於使共軛二烯(共)聚合的工藝、特別地使1,3-丁二烯聚合的工藝,其特徵在於工藝使用所述催化體系。
已知的是,共軛二烯的立體定向(共)聚合(stereospecific(co)polymerization)在用於獲得最廣泛使用的橡膠中的產品的化學工業中是非常重要的工藝。
所述立體定向(共)聚合可以給出具有不同結構的聚合物,也就是,1,4-反式結構、1,4-順式結構、1,2結構,以及在不對稱共軛二烯(例如,異戊二烯)的情況下,3,4結構。如果沿著聚合物鏈存在不對稱碳原子,如例如在衍生自1,3-戊二烯的聚合的聚合物的情況下,具有1,4-順式結構和1,4-反式結構的有規立構聚合物還可以是全同立構的或間同立構的。具有1,2結構或3,4結構的有規立構聚合物也可以是全同立構的或間同立構的,並且取決於聚合的共軛二烯的結構,在聚合物側鏈中的雙鍵可以具有1,4-反式結構或1,4-順式結構。
前面提及的有規立構聚合物僅可以通過使用齊格勒-納塔型的催化體系的立體定向聚合來獲得,所述齊格勒-納塔型的催化體系通常通過使過渡金屬或鑭系化合物比如例如滷化物、醇化物、羧酸鹽、具有各種類型的配體的有機金屬化合物,與合適的烷基化劑例如烷基鋁[例如,Al(R)3,其中R可以是例如甲基、乙基、正丙基、異丙基、正丁基、異丁基]、或鋁氧烷(aluminoxane)[例如,甲基鋁氧烷(MAO)]組合來獲得。這是因為與其他聚合的方法(例如,自由基聚合、負離子聚合)相比,立體定向聚合以如下為特徵:(i)高區域選擇性,也就是,其能夠提供由單類型的結構(也就是,1,4結構、或1,2結構、或3,4結構)形成的聚合物;(ii)高立體選擇性,也就是,如果在共軛二烯中存在空間異構位點(steric isomerism site)(例如,內部雙鍵、不對稱碳原子),那麼其能夠提供具有高構型順序(configurational order)的聚合物。關於所述立體定向聚合的另外的細節可以在例如以下中被找到:Porri L.等人,「Comprehensive Polymer Science」(1989),Eastmond G.C.等人編輯,Pergamon Press,Oxford,UK,第4卷,第II部分,第53頁-第108頁;Thiele S.K.H.等人,「Macromolecular Science.Part C:Polymer Reviews」(2003),C43,第581頁-第628頁;Osakada,K.等人,「Advanced Polymer Science」(2004),第171卷,第137頁-第194頁;Ricci G.等人,「Advances in Organometallic Chemistry Research」(2007),Yamamoto K.編輯,Nova Science Publisher,Inc.,USA,第1頁-第36頁;Ricci G.等人,「Coordination Chemistry Reviews」(2010),第254卷,第661頁-第676頁;Friebe L.等人,「Advanced Polymer Science」(2006),第204卷,第1頁-第154頁。
還已知的是,前面提及的有規立構聚合物特別是聚丁二烯和聚異戊二烯的特徵和應用取決於所述聚合物的微觀結構而顯著變化。因此,這些在從用於製備用於輪胎生產的摻合物的、其特徵是非常低的玻璃化轉變溫度(Tg)(對於聚丁二烯,約-110℃)的典型的彈性聚合物(即,具有高1,4-順式含量的聚丁二烯和聚異戊二烯),到主要用於鞋底的生產並且其特徵是相對高的熔點(Tm)(對於1,2間同立構的聚丁二烯,約220℃)的結晶聚合物(即,1,2間同立構的聚丁二烯和3,4間同立構的聚異戊二烯)的範圍內。
使用基於過渡金屬的催化體系,共軛二烯的立體定向聚合開始於1954年,就在丙烯的聚合中獲得的第一次結果之後。使用的第一種催化體系通過使四氯化鈦(TiCl4)或三氯化鈦(TiCl3)與烷基鋁組合來獲得,也就是先前被用於使乙烯或丙烯聚合的催化體系。
第一種合成的有規立構二烯聚合物是聚異戊二烯,其具有由Horne S.E.等人在「Industrial&Engineering Chemistry」(1956),第48(4)卷,第784頁-第791頁中描述的、非常類似於天然橡膠的結構的結構(也就是,1,4-順式結構),隨後很快是,具有由Natta G.等人在「Chemical Abstract」(1959),第53卷,第3756頁中和在義大利專利申請IT 536631中描述的、類似於古塔波膠的結構的結構(也就是,1,4-反式結構)的聚異戊二烯。
60年代初之前,所有聚丁二烯的立體異構體已經被合成,也就是,1,4-順式、1,4-反式、1,2-間同立構的和1,2-全同立構的立體異構體。隨後,通過使過渡金屬[例如,鈦(Ti)、釩(V)、鉻(Cr)、鐵(Fe)、鈷(Co)、鎳(Ni)]或鑭系元素[例如,釹(Nd)、鐠(Pr)、釓(Gd)、鑭(La)]的化合物(例如,滷化物、醇化物、羧酸鹽)與合適的烷基化劑[例如,三乙基鋁(AlEt3)、氯化二乙基鋁(AlEt2Cl)]組合而獲得的各種其他催化體系被提議並且研究。
還已知的是,在從1,3-丁二烯(即,1,4-順式、1,4-反式、1,2間同立構、1,2全同立構、1,2無規立構、具有可變的1,2單元含量的混合的l,4-順式/l,2結構)的立體定向聚合可獲得的多種聚合物中,僅僅1,4-順式聚丁二烯和1,2間同立構聚丁二烯在工業上被生產且商業化。
具有高1,4-順式含量的聚丁二烯是通常具有96%-97%的1,4-順式單元的含量、約-2℃的熔點(Tm)、約-25℃的結晶點(Tc)和小於-100℃的玻璃化轉變溫度的合成彈性體,所述合成彈性體的性質非常類似於天然橡膠的性質,並且所述合成彈性體的主要用途是在彈性摻合物,特別是用於汽車和/或卡車的輪胎的生產的彈性摻合物的生產中。特別地,在輪胎生產中,使用具有高的1,4-順式單元含量的聚丁二烯。通常,1,4-順式聚丁二烯通過聚合工藝來製備,所述聚合工藝使用基於鈦(Ti)、鈷(Co)、鎳(Ni)、釹(Nd)的多種催化體系。
1,2間同立構聚丁二烯是可溶性差的結晶聚合物,其具有在從200℃至220℃的範圍內的熔點,這取決於間同立構規整度的水平而變化(換句話說,取決於包含在其中的間同立構五價基(syndiotactic pentad)的百分比),並且通常被用於生產透明膜、軟管,特別地用於生產鞋底。
因此,存在用於1,3-丁二烯的聚合的許多催化體系。
例如,基於釩(V)的催化體系在共軛二烯的聚合領域中已知的是其提供具有1,4-反式結構的聚合物的能力,並且到目前為止是用於製備1,4-反式聚丁二烯的最重要的體系。關於所述催化體系的另外的細節可以在例如以下中被找到:上文引用的Porri L.等人,「Comprehensive Polymer Science」(1989),Eastmond G.C.等人編輯,Pergamon Press,Oxford,UK,第4卷,第II部分,第53頁-第108頁。
通過使釩的滷化物[例如,三氯化釩(VCl3)、四氯化釩(VCl4)]與烷基鋁[例如,三乙基鋁(AlEt3)、氯化二乙基鋁(AlEt2Cl)]組合獲得的非均相催化體系(heterogeneous catalytic system)提供具有約145℃的熔點(Tm)的、高分子量的、結晶1,4-反式聚丁二烯(97%-100%的1,4-反式含量)。關於所述催化體系的另外的細節可以在例如以下中被找到:Natta G.等人,「La Chimica e L』Industria」(1959),第40卷,第362頁和「Chemical Abstract」(1959),第53卷,第195頁;Natta G.等人,「La Chimica e L』Industria」(1959),第41卷,第116頁和「Chemical Abstract」(1959),第53卷,第15619頁。
具有高的1,4-反式單元含量但具有較低分子量的聚丁二烯可以使用均相催化體系比如例如氯化釩(III)(三-四氫呋喃(tris-tetrahydrofuran))/氯化二乙基鋁(VCl3(THF)3/AlEt2Cl)、乙醯丙酮釩(III)/氯化二乙基鋁[V(acac)3/AlEt2Cl]和乙醯丙酮釩(III)/甲基鋁氧烷[V(acac)3/MAO]來製備。關於所述催化體系的另外的細節可以在例如以下中被找到:Ricci G.等人,「Polymer Communication」(1991),第32卷,第514頁-第517頁;Ricci G.等人,「Journal of Polymer Science Part A:Polymer Chemistry」(2007),第45卷,第4635頁-第4646頁;Natta G.等人,「Atti Accademia Nazionale dei Lincei-Classe di Scienze fisiche,matematiche e naturali」(1961),第31(5)卷,第189頁和「Chemical Abstract」(1962),第57卷,第4848頁;Porri L.等人,「Die Makromolekulare Chemie」(1963),第61(1)卷,第90頁-第103頁。
前面提及的均相催化體系中的某些,例如乙醯丙酮釩(III)/三乙基鋁[V(acac)3/AlEt3]對於製備1,2-丁二烯受到某些關注,如例如在Natta G.等人,「La Chimica e L』Industria」(1959),第41卷,第526頁和「Chemical Abstract」(1960),第54卷,第1258頁中所描述的。
通過使釩的環戊二烯基衍生物比如例如氯化雙(環戊二烯基)釩(Cp2VCl)和二氯化甲基環戊二烯基釩雙三乙基膦[(C5H4Me)VCl2(PEt3)2]組合獲得的催化體系能夠提供具有主要地1,4-順式結構的聚丁二烯(約85%的1,4-順式單元含量)。關於所述催化體系的另外的細節可以在例如以下中被找到:Porri L.等人,「Metalorganic Catalyst for Synthesis and Polymerization」(1999),Kaminsky W.編輯,Springer-Verlag Berlin Heidelberg,第519頁-第530頁;Porri L.等人,「Metallocene-Based Polyolefins」(2000),Scheirs J.等人編輯,John Wiley&Sons Ltd.,第115頁-第141頁;Natta G.編輯,「Atti Accademia Nazionale dei Lincei-Classe di Scienze fisiche,matematiche e naturali」(1961),第31(5)卷,第189頁和「Chemical Abstract」(1962),第57卷,第4848頁;Porri L.等人,「Die Makromolekulare Chemie」(1963),第61(1)卷,第90頁-第103頁。
基於鉻的催化體系在使共軛二烯聚合的領域中起重要作用,其已經是能夠提供具有1,2結構的聚丁二烯的首要的催化體系之一。例如,通過使可溶性鉻化合物比如例如乙醯丙酮鉻(III)[Cr(acac)3]或五羰基吡啶鉻[Cr(CO)5吡啶]與烷基鋁[例如三乙基鋁(AlEt3)]組合獲得的催化體系已經使得獲得具有全同立構或間同立構的結構的1,2聚丁二烯是可能的,取決於使用的Al/Cr摩爾比:在低Al/Cr比率下為間同立構的,即在從2至6的範圍內的比率;在高Al/Cr比率下為全同立構的,即在從6至10的範圍內的比率,如例如在Natta G.等人,「La Chimica e L』Industria」(1959),第41卷,第1163頁中所描述的。明顯的並且確證其重要性的是,使用基於鉻的催化體系之前不是僅獲得全同立構的1,2聚丁二烯。
最近這些年,新的、更有活性的且立體專一性的催化體系已經通過使鉻(II)[Cr(II)]和二齒次膦酸配體(bidentate phosphinic ligand)的各種絡合物與甲基鋁氧烷(MAO)組合來開發,如例如在:Ricci G.等人,「Chromium:Environmental,Medical and Material Studies」(2011),Salden M.P.編輯,Nova Science Publishers Inc.,USA,第121頁-第140頁;Ricci G.等人,「Macromolecules」(2001),第34卷,第5766頁-第5769頁;Ricci G.等人,「Polymer Bullettin」(2002),第48卷,第25頁-第31頁;Ricci G.等人,「Organometallics」(2004),第23(15)卷,第3727頁-第3732頁;Ricci G.等人,「Journal of Molecular Catalysis A:Chemical」(2007),第267卷,第102頁-第107頁;Ricci G.等人,「Macromolecular Symposia」(2004),第260(1)卷,第172頁-第178頁中所描述的。所述催化體系已經使得獲得具有多達95%的1,2單元含量,具有不同的立構規整度也就是全同立構規整度或間同立構規整度(取決於與鉻原子配位的膦的類型)的聚丁二烯是可能的。特別地,主要地全同立構聚合物已經使用較小空間位阻的膦[例如雙(二甲基膦)甲烷(dmpm)、雙(二苯基膦)甲烷(dppm)]獲得,同時使用較大空間位阻的膦[例如1,2-雙(二甲基膦)乙烷(dmpe)、1,2-雙(二乙基膦)乙烷(depe)、雙(二苯基膦)胺(dppa)、1,2-雙(二苯基膦)乙烷(dppe)]已經使得合成高度間同立構的1,2聚丁二烯是可能的。
同時,通過與其他過渡金屬例如鈦(Ti)、釩(V)、鉻(Cr)、鈷(Co)和鎳(Ni)相比,基於鐵(Fe)的催化體系一直相對少地被研究。然而,已經獲得極其有活性的催化體系,即使它們不具有高立體專一性的特徵。例如,對於基於二氯化鐵或二乙基鐵與芳族胺(例如,菲咯啉、聯吡啶)和烷基鋁[例如,三異丁基鋁(Al(iBu)3)、三乙基鋁(AlEt3)、甲基鋁氧烷(MAO)]的絡合物的催化體系就是這種情況,所述催化體系通過使1,3-丁二烯聚合,在非常短的時間內在單體(1,3-丁二烯)的完全轉化下提供主要地1,2聚丁二烯(~70%;其餘單元是1,4-順式類型)。關於所述催化體系的另外的細節可以在例如以下中被找到:Bazzini C.等人,「Macromolecular Rapid Communications」(2002),第23(15)卷,第922頁-第927頁;Ricci G.等人,「Journal of Molecular Catalysis A:Chemical」(2003),第204卷-第205卷,第287頁-第293頁;Bazzini C.等人,「Polymer」(2004),第45卷,第2871頁-第2875頁;Ricci G.等人,「Ferrocenes:Compounds,Properties and Applications」(2011),Phillips E.S.編輯,Nova Science Publishers Inc.,USA,第273-314頁。
基於鈷的催化體系可能是用於使共軛二烯聚合的、基於過渡金屬的各種催化體系中最通用的催化體系,由於在其催化配方(catalytic formulation)中合適的變形的情況下,它們能夠提供,在呈現高活性和立體專一性下,全部可能是1,3-丁二烯的立體異構體:1,4-順式聚丁二烯、1,2聚丁二烯、具有混合的1,4-順式/1,2結構的聚丁二烯、1,4反式聚丁二烯。關於所述立體定向聚合的另外的細節可以在例如以下中被找到:Porri L.等人,「Comprehensive Polymer Science」(1989),Eastmond G.C.等人編輯,Pergamon Press,Oxford,UK,第4卷,第II部分,第53頁-第108頁;Thiele S.K.H.等人,「Macromolecular Science.Part C:Polymer Reviews」(2003),C43,第581頁-第628頁;Osakada,K.等人,「Advanced Polymer Science」(2004),第171卷,第137頁-第194頁;Ricci G.等,「Advances in Organometallic Chemistry Research」(2007),Yamamoto K.編輯,Nova Science Publisher,Inc.,USA,第1頁-第36頁;以及Ricci G.等人,「Coordination Chemistry Reviews」(2010),第254卷,第661頁-第676頁;上文所列出的;以及Ricci G.等人,「Cobalt:Characteristics,Compounds,and Applications」(2011),Lucas J.Vidmar編輯,Nova Science Publisher,Inc.,USA,第39頁-第81頁。
乙醯丙酮鈷(II)/氯化二乙基鋁/水[Co(acac)2/AlEt2Cl/H2O]和乙醯丙酮鈷(III)/氯化三乙基鋁/水/二硫化碳[Co(acac)3/AlEt3/H2O/CS2]催化體系仍舊分別被用於1,4-順式聚丁二烯和間同立構的1,2聚丁二烯的工業生產。
在最近這些年,新的催化體系已經通過使二氯化鈷(CoCl2)絡合物與膦配體組合來獲得,如例如在:Ricci G.等人,「Journal of Molecular Catalysis A:Chemical」(2005),第226卷,第235頁-第241頁;Ricci G.等人,「Macromolecules」(2005),第38卷,第1064頁-第1070頁;Ricci G.等人,「Journal of Organometallic Chemistry」(2005),第690卷,第1845頁-第1854頁;Takeuchi M.等人,「Polymer International」(1992),第29卷,第209頁-第212頁;Takeuchi M.等人,「Polymer International」(1995),第36卷,第41頁-第45頁;Takeuchi M.等人,「Macromolecular Chemistry and Physics」(1996),第197卷,第729頁-第743頁中所描述的。
前面提及的新的催化體系的特性是,它們使得通過改變與鈷配位的配體的類型獲得具有受控制的微結構(1,4-順式結構、1,2結構、混合的1,4-順式/1,2結構)的聚丁二烯是可能的。例如,具有高的1,4-順式單元含量的聚丁二烯已經在受阻脂肪族膦(hindered aliphatic phosphine)[例如三叔丁基膦(PtBu3)、三異丙基膦(PiPr3)]的情況下獲得,同時具有混合的1,4-順式/1,2結構的聚丁二烯已經使用具有較小空間位阻的脂肪族膦[例如三乙基膦(PEt3)、三正丙基膦(PnPr3)]獲得,如例如在Ricci G.等人,「Cobalt:Characteristics,Compounds,and Applications」(2011),Lucas J.Vidmar編輯,Nova Science Publishers,Inc.,USA,第39頁-第81頁;Ricci G.等人,「Journal of Molecular Catalysis A:Chemical」(2005),第226卷,第235頁-第241頁;上文所引用的中所描述的。
同時,使用基於具有芳族膦的絡合物的催化體系已經導致形成主要地1,2聚丁二烯,隨膦的空間位阻增大,其具有的間同立構規整度的水平增大。
多年以來,多種基於鎳的催化體系一直被用於1,3-丁二烯的聚合,例如環烷酸鎳(II)/氯化二乙基鋁/水[Ni(環烷酸鹽)2/AlEt2Cl/H2O]、環戊二烯基鎳(II)/甲基鋁氧烷[NiCp2/MAO]、乙醯丙酮鎳(II)/甲基鋁氧烷[Ni(acac)2/MAO],如例如在:Porri L.等人,「Comprehensive Polymer Science」(1989),Eastmond G.C.等人編輯,Pergamon Press,Oxford,UK,第4卷,第II部分,第53頁-第108頁;Thiele S.K.H.等人,「Macromolecular Science.Part C:Polymer Reviews」(2003),C43,第581頁-第628頁;Osakada K.等人,「Advanced Polymer Science」(2004),第171卷,第137頁-194頁;上文所引用的;以及在:Oliva P.等人,「Die Makromolekulare Chemie,Rapid Communications」(1990),第11(11)卷,第519頁-第524頁;Sato H.等人,「Bulletin of the Chemical Society of Japan」(1992),第65卷,第5期,第1299頁-第1306頁;Longo P.等人,「Macromolecular Rapid Communications」(1998),第19(1)卷,第31頁-第34頁中所描述的。
相比於基於鈷的催化體系的那些,前面提及的基於鎳的催化體系中的某些具有活性和立體專一性,並且具有工業興趣。特別地,三乙基鋁/辛酸鎳(II)/三氟化硼二乙醚[Al/Et3/Ni(辛酸鹽)2/BF3·OEt2]催化體系當前被用於具有高1,4-順式含量(即,96%-97%的1,4-順式單元含量)的工業生產,如例如在德國專利DE 2,113,527和在Throckmorton M.C.等人,「Rubber Chemistry and Technology」(1972),第45卷,第268頁-第277頁;Saltman W.等人,「Rubber Chemistry and Technology」(1973),第46卷,第1055頁-第1067頁中所描述的。
另外,對於基於鑭的催化體系還已知的是其不僅在1,3-丁二烯而且在許多其他被取代的丁二烯的1,4-順式聚合中的高專一性,如例如在以下中所描述的:Porri L.等人,「Comprehensive Polymer Science」(1989),Eastmond G.C.等人編輯,Pergamon Press,Oxford,UK,第4卷,第II部分,第53頁-第108頁;Osakada,K.等人,「Advanced Polymer Science」(2004),第171卷,第137頁-第194頁;Ricci G.等人,「Advances in Organometallic Chemistry Research」(2007),Yamamoto K.編輯,Nova Science Publisher,Inc.,USA,第1頁-第36頁;Ricci G.等人,「Coordination Chemistry Reviews」(2010),第254卷,第661頁-第676頁;Friebe L.等人,「Advanced Polymer Science」(2006),第204卷,第1頁-第154頁;上文所引用的。
基於釹(Nd)、釓(Gd)和鐠(Pr)的催化體系由中國研究者在60年代早期被研究,如例如在Hsieh L.等人,「Rubber Chemistry and Technology」(1972),第45卷,第268頁中所描述的,並且立刻就發現相對於被用於1,4-順式聚丁二烯的合成的其他催化體系具有某些優點。特別地,所述催化體系提供具有比使用基於鈷(Co)、鎳(Ni)和鈦(Ti)的催化體系獲得的那些更線型(linear)的1,4-順式結構的聚丁二烯,並且因此更適於輪胎生產,其是目前為止1,4-順式聚丁二烯的最重要的實際應用。
包含基於釹的化合物的常規催化體系通過使釹化合物比如例如乙醯丙酮釹(III)[Nd(acac)3]、2-乙基己酸釹(III)[Nd(OCOC7H15)3]與氯供體比如例如氯化二乙基鋁(AlEt2Cl)、乙基-倍半氯化鋁(ethyl-aluminium sesquichloride)(Al2Et3Cl3)、叔丁基氯並且與烷基鋁例如三異丁基鋁(AliBu3)、二異丁基氫化鋁(AliBu2H)反應來獲得。所述催化體系當前被用於具有非常高1,4-順式含量,即98%的1,4-順式單元含量的聚丁二烯的工業生產。關於所述催化體系的另外的細節可以在例如以下中被找到:上文引用的Friebe L.等人,「Advanced Polymer Science」(2006),第204卷,第1頁-第154頁;Cabassi F.等人,「Transition Metal Catalyzed Polymerizations」(1988),Quirk R.P.編輯,Cambridge University Press,MA,USA,第655頁;Ricci G.等人,「Polymer Communication」(1987),第28卷,第223頁;Wilson D.J.等人,「Polymer Bulletin」(1992),第27卷,第407頁-第411頁;Porri L.等人,「Macromolecular Symposia」(1998),第128(1)卷,第53頁-第61頁;Porri L.等人,「ACS Symposium Sereies」(2000),第749頁,第15頁-第30頁。
氯化鈦(IV)/三烷基鋁催化體系(TiCl4/Al(R)3,其中R可以是例如甲基、乙基、異丁基、環己基)是被用於使1,3-丁二烯聚合的第一種催化劑,如例如在以下中所描述的:Porri L.等人,「Comprehensive Polymer Science」(1989),Eastmond G.C.等人編輯,Pergamon Press,Oxford,UK,第4卷,第II部分,第53-108頁;Horne S.E.等人,「Industrial Engineering Chemistry」(1956),第48卷,第784-791頁;上文所引用的。取決於Al/Ti摩爾比,可以獲得主要具有1,4-順式結構的聚丁二烯(即,65%-70%的1,4-順式單元含量)或具有混合的1,4-順式/1,4-反式結構的聚丁二烯。
具有約92%-95%的較高的1,4-順式單元含量的聚丁二烯已經通過使多種類型的鋁的烷基化合物比如例如式Al(R)3的化合物,其中R可以是例如甲基、乙基、異丁基、環己基,優選地三異丁基鋁[Al(iBu)3],與包含碘的基於鈦的催化體系(例如碘化鈦(IV)(TiI4)、二碘二氯化鈦(TiCl2I2)、三氯碘化鈦(TiCl3I))組合來獲得,如例如在以下中所描述的:Porri L.等人,「Comprehensive Polymer Science」(1989),Eastmond G.C.等人編輯,Pergamon Press,Oxford,UK,第4卷,第II部分,第53頁-第108頁,上文所引用的;Cooper W.等人,「The Stereo Rubbers」(1997),W.M.Saltman編輯,Wiley,New York,第21頁;Marconi W.等人,「La Chimica e l』Industria」(1963),第45卷,第522頁-第528頁;Marconi W.等人,「Journal of Polymer Science Part A:General Papers」(1965),第3(2)卷,第735頁-第752頁。
基於鈦的催化體系是被用於合成具有高的1,4-順式單元含量的聚丁二烯的第一種催化體系,並且在歐洲和美國兩者,充當用於開發工業上用於所述合成的工藝的基礎。當今,更多有活性和立體專一性的催化體系是可用的,其基於其他金屬比如例如鈷(Co)、鎳(Ni)和釹(Nd)。
然而,在催化配方的合適的變形下,基於鈦的催化體系也能夠提供具有1,2結構和1,4-反式結構的聚丁二烯。例如,α-三氯化鈦/三乙基鋁(α-TiCl3/AlEt3)催化體系是被用於製備1,4-反式聚丁二烯的第一種催化劑,如例如在以下中所描述的:Porri L.等人,「Comprehensive Polymer Science」(1989),Eastmond G.C.等人編輯,Pergamon Press,Oxford,UK,第4卷,第II部分,第53頁-第108頁;Natta G.等人在「Chemical Abstract」(1959),第53卷,第3756頁和在義大利專利申請IT 536631;上文所引用的。
通過與基於鈦的催化體系相比,基於鋯的催化體系一直被研究非常少,可能是因為基於鋯的催化體系被認為對於使共軛二烯聚合的有效性差。然而,最近,能夠提供具有高的1,4-順式單元含量(即,1,4-順式單元含量≥99.9%)的聚丁二烯的、基於鋯的吡啶絡合物的新的催化劑已經被描述:關於所述催化劑的進一步的細節可以在Annunziata L.等人,「Macromolecules」(2011),第44卷,第1934頁-第1941頁中被找到。
由於具有高的1,4-反式單元含量的共軛二烯的(共)聚合物,特別地聚丁二烯,可以有利地被用於生產輪胎,特別地具有良好的耐磨性的輪胎的胎面,以及鞋類工業(例如,鞋底的生產)中,所以能夠提供所述(共)聚合物的新的催化體系的研究仍舊具有很大的興趣。
本申請人已經為自己設定以下任務:找到可以被用於催化體系的新穎的鋯的絡合物,該催化體系能夠提供具有高的1,4-反式單元含量,即1,4-反式單元含量≥94%的共軛二烯的(共)聚合物例如比如直鏈的或支鏈的聚丁二烯。
現在,本申請人已經找到具有如下文定義的通式(I)的新穎的鋯的吡啶絡合物,其能夠提供具有高的1,4-反式單元含量,即1,4-反式單元含量≥94%的共軛二烯的(共)聚合物比如例如直鏈的或支鏈的聚丁二烯。
因此,本發明涉及具有通式(I)的鋯的吡啶絡合物:
其中:
-相同或不同的R1和R2代表氫原子;或選自直鏈的或支鏈的、任選地被滷化的C1-C20、優選地C1-C15烷基,任選地被取代的環烷基,任選地被取代的芳基;
-相同或不同的R3、R4、R5和R6代表氫原子;或選自直鏈的或支鏈的、任選地被滷化的C1-C20、優選地C1-C15烷基,任選地被取代的環烷基,任選地被取代的芳基,硝基,羥基,氨基;
-相同或不同的X1、X2和X3代表滷素原子例如比如氯、溴、碘、優選地氯;或選自直鏈的或支鏈的C1-C20、優選地C1-C15烷基,-OCOR7基團或-OR7基團,其中R7選自直鏈的或支鏈的C1-C20、優選地C1-C15烷基;或X1、X2和X3中的一個代表具有通式(II)的基團:
其中R1、R2、R3、R4、R5和R6具有上文描述的相同的含義。
為了本說明書和以下的權利要求的目的,除非另有陳述,否則數值範圍的定義總是包括端點。
為了本說明書和以下的權利要求的目的,術語「包含(comprising)」還包括術語「大體上由...組成」或「由...組成」。
術語「C1-C20烷基」指的是具有從1個到20個碳原子的直鏈的或支鏈的烷基。C1-C20烷基的特定的實例是:甲基、乙基、正丙基、異丙基、正丁基、仲丁基、異丁基、叔丁基、戊基、己基、庚基、辛基、正壬基、正癸基、2-丁基辛基、5-甲基己基、4-乙基己基、2-乙基庚基、2-乙基己基。
術語「任選地被滷化的C1-C20烷基」指的是飽和或不飽和的、具有從1個到20個碳原子的直鏈的或支鏈的烷基,其中至少一個氫原子被滷素原子比如例如氟、氯、溴(優選地氟、氯)取代。任選地被滷化的C1-C20烷基的特定的實例是:氟甲基、二氟甲基、三氟甲基、三氯甲基、2,2,2-三氟乙基、2,2,2-三氯乙基、2,2,3,3-四氟丙基、2,2,3,3,3-五氟丙基、全氟戊基、全氟辛基、全氟癸基。
術語「環烷基」指的是具有從3個到30個碳原子的環烷基。所述環烷基可以任選地被選自以下的相同或不同的一個或更多個基團取代:滷素原子、羥基、C1-C12烷基、C1-C12烷氧基、氰基、氨基、硝基。環烷基的特定的實例是:環丙基、2,2-二氟環丙基、環丁基、環戊基、環己基、六甲基環己基、五甲基環戊基、2-環辛基乙基、甲基環己基、甲氧基環已基、氟環己基、苯基環己基。
術語「芳基」指的是芳香族碳環基團。所述芳香族碳環基團可以任選地被選自以下的相同或不同的一個或更多個基團取代:滷素原子,比如例如氟、氯、溴;羥基;C1-C12烷基;C1-C12烷氧基;氰基;胺基;硝基。芳基的特定的實例是:苯基、甲基苯基、三甲基苯基、甲氧基苯基、羥基苯基、苯氧基苯基、氟苯基、五氟苯基、氯苯基、溴苯基、硝基苯基、二甲基氨基苯基、萘基、苯基萘基、菲、蒽。
在本發明的優選的實施方案中,在所述具有通式(I)的鋯的吡啶絡合物中:
-相同或不同的R1和R2代表氫原子;或選自C1-C20烷基,優選地是甲基、任選地被取代的芳基,優選地是苯基、或被一個或更多個甲基、異丙基、叔丁基取代的苯基;優選地R1是氫原子或甲基並且R2是苯基、或被一個或更多個甲基、異丙基、叔丁基取代的苯基;
-彼此相同的R3、R4、R5和R6代表氫原子;
-相同或不同的X1、X2和X3代表滷素原子,比如例如氯、溴、碘,優選地氯;或X1、X2和X3中的一個代表具有通式(II)的基團:
其中R1、R2、R3、R4、R5和R6具有上文描述的相同的含義。
根據本發明,具有通式(I)的鋯的吡啶絡合物應被認為是呈任何物理形式,比如例如分離和純化的固體的形式,被合適的溶劑溶劑化的形式,或被負載在合適的有機固體或無機固體上的形式,優選地具有顆粒或粉末的物理形式。
具有通式(I)的鋯的吡啶絡合物從具有通式(III)的配體來製備:
其中R1、R2、R3、R4、R5和R6具有上文描述的相同的含義。
可用於本發明的目的的配體的特定的實例是具有以下式(L1)-(L7)的那些:
所述具有式(L1)-(L7)的配體可以通過本領域已知的工藝來製備。例如,所述具有式(L1)-(L7)的配體可以通過包括以下的工藝來製備:(1)在合適的苯胺和2-吡啶甲醛(2-pyridincarboxyaldehyde)或2-乙醯基吡啶之間的縮合反應,形成對應的亞胺,如例如在Wu J.等人,「Journal of American Chemistry Society」(2009),第131(36)卷,第12915頁-第12917頁;Laine V.T.等人,「European Journal of Inorganic Chemistry」(1999),第6卷,第959頁-第964頁;Bianchini C.等人,「New Journal of Chemistry」(2002),第26(4)卷,第387頁-第397頁;Lai Yi-C.等人,「Tetrahedron」(2005),第61(40)卷,第9484頁-第9489頁中所描述的;(2)合成的亞胺轉化成對應的胺,如例如在:Nienkemper K.等人,「Journal of Organometallic Chemistry」(2008),第693(8-9)卷,第1572頁-第1589頁;Lin Y.等人,「Dalton Transactions」(2012),第41(22)卷,第6661頁-第6670頁中所描述的。
具有通式(I)的鋯的吡啶絡合物可以通過本領域已知的工藝來製備。例如,所述鋯的吡啶絡合物可以通過在原樣的或與醚[例如,二乙醚、四氫呋喃(THF)、二甲氧基乙烷]絡合的、具有通式Zr(X)4的鋯化合物(其中X是滷素原子比如例如氯、溴、碘,優選地氯),與具有如上文陳述的式(L1)-(L7)的配體(所述配體以化學計量的量來使用)之間,優選地在至少一種溶劑的存在下,在從25℃至110℃的範圍內的溫度、優選地在溶劑的回流溫度下操作的反應來製備,所述至少一種溶劑可以選自例如:氯化溶劑(例如,二氯甲烷)、醚溶劑[例如,四氫呋喃(THF)]、烴溶劑(例如,甲苯)、或其混合物。另外,如果X1、X2和X3中的一個代表具有通式(II)的基團
其中R1、R2、R3、R4、R5和R6具有上文描述的相同的含義,那麼在與前面提及的鋯化合物反應之前,使所述配體與烷基鋰比如例如正丁基鋰(n-BuLi)反應,以獲得所述配體的鹽,所述配體的鹽隨後與前面提及的鋯化合物在如上文描述操作下反應。如此獲得的鋯的吡啶絡合物可以隨後通過本領域已知的方法來回收,比如例如使用非溶劑(例如,戊烷)的沉澱、隨後通過過濾或傾析的分離和任選的隨後的溶解於合適的溶劑中、隨後低溫結晶。
為了本說明書和以下的權利要求書的目的,措辭「室溫」指的是在從20℃到25℃的範圍內的溫度。
如先前陳述的,本發明還涉及包含所述具有通式(I)的鋯的吡啶絡合物的用於使共軛二烯(共)聚合的催化體系。
因此,本發明還涉及用於使共軛二烯(共)聚合的催化體系,所述催化體系包含:
(a)至少一種具有通式(I)的鋯的吡啶絡合物;
(b)至少一種助催化劑,所述至少一種助催化劑選自不同於碳的元素M'的有機化合物,所述元素M'選自屬於元素周期表的第2族、第12族、第13族或第14族的元素,優選地選自:硼、鋁、鋅、鎂、鎵、錫,還更優選地選自鋁、硼。
通常,包含具有通式(I)的鋯的吡啶絡合物和助催化劑(b)的催化體系的形成優選地在惰性液體介質中、更優選地在烴溶劑中進行。對具有通式(I)的鋯的吡啶絡合物和助催化劑(b)以及使用的特定方法的選擇可以取決於分子結構和期望的結果而變化,如在用於其他的過渡金屬與亞胺配體的絡合物的在本領域技術人員可用的有關文獻中類似地報告的,如例如由L.K.Johnson等在「Journal of the American Chemical Society」(1995),第117卷,第6414頁-第6415頁中和G.van Koten等在「Advances in Organometallic Chemistry」(1982),第21卷,第151頁-第239頁中報告的。
在本發明的另外的優選的實施方案中,所述助催化劑(b)可以選自具有通式(IV)的烷基鋁(b1):
Al(X')n(R8)3-n(IV)
其中X'代表滷素原子比如例如氯、溴、碘、氟;R8選自直鏈的或支鏈的C1-C20烷基、環烷基、芳基,所述基團任選地被一個或更多個矽原子或鍺原子取代;並且n是在從0到2的範圍內的整數。
在本發明的另外的優選的實施方案中,所述助催化劑(b)可以選自屬於元素周期表的第13族或第14族的不同於碳的元素M'的有機氧化化合物(organo-oxygenated compound)(b2),優選地鋁、鎵、錫的有機氧化化合物。所述有機氧化化合物(b2)可以被定義為M'的有機化合物,其中此M'被結合到至少一個氧原子並且被結合到由具有從1個到6個碳原子的烷基、優選地甲基形成的至少一個有機基團。
在本發明的另外的優選的實施方案中,所述助催化劑(b)可以選自不同於碳的元素M'的有機金屬化合物或有機金屬化合物的混合物(b3),所述有機金屬化合物或有機金屬化合物的混合物(b3)能夠與具有通式(I)的鋯的吡啶絡合物反應,從具有通式(I)的鋯的吡啶絡合物中選取(extract)σ-連接的取代基X1、X2或X3,以在一方面形成至少一種中性化合物,且在另一方面形成由包含被配體配位的金屬(Zr)的陽離子和包含金屬M'的非配位的有機陰離子組成的離子化合物,離子化合物的負電荷被離域在多中心結構上。
應當注意到的是,為了本發明和以下權利要求的目的,術語「元素周期表」指的是在以下網際網路網址可用的日期為2007年6月22日的版本的「IUPAC元素周期表」:www.iupac.org/fileadmin/user_upload/news/IUPAC_Periodic_Table-1Jun12.pdf。
對本發明的目的特別有用的具有通式(IV)的烷基鋁的特定的實例是:三甲基鋁、三(2,3,3-三甲基-丁基)-鋁、三(2,3-二甲基-己基)-鋁、三(2,3-二甲基-丁基)-鋁、三(2,3-二甲基-戊基)-鋁、三(2,3-二甲基-庚基)-鋁、三(2-甲基-3-乙基-戊基)-鋁、三(2-甲基-3-乙基-己基)-鋁、三(2-甲基-3-乙基-庚基)-鋁、三(2-甲基-3-丙基-己基)-鋁、三乙基鋁、三(2-乙基-3-甲基-丁基)-鋁、三(2-乙基-3-甲基-戊基)-鋁、三(2,3-二乙基-戊基)-鋁、三正丙基鋁、三異丙基鋁、三(2-丙基-3-甲基-丁基)-鋁、三(2-異丙基-3-甲基-丁基)-鋁、三正丁基鋁、三異丁基鋁(TIBA)、三叔丁基鋁、三(2-異丁基-3-甲基-戊基)-鋁、三(2,3,3-三甲基-戊基)-鋁、三(2,3,3-三甲基-己基)-鋁、三(2-乙基-3,3-二甲基-丁基)-鋁、三(2-乙基-3,3-二甲基-戊基)-鋁、三(2-異丙基-3,3-二甲基-丁基)-鋁、三(2-三甲基甲矽烷基-丙基)-鋁、三(2-甲基-3-苯基-丁基)-鋁、三(2-乙基-3-苯基-丁基)-鋁、三(2,3-二甲基-3-苯基-丁基)-鋁、三(2-苯基-丙基)-鋁、三[2-(4-氟-苯基)-丙基]-鋁、三[2-(4-氯-苯基)-丙基]-鋁、三-[2-(3-異丙基-苯基)-三(2-苯基-丁基)]-鋁、三(3-甲基-2-苯基-丁基)-鋁、三(2-苯基-戊基)-鋁、三[2-(五氟-苯基)-丙基]-鋁、三(2,2-二苯基-乙基]-鋁、三(2-苯基-甲基-丙基]-鋁、三戊基鋁、三己基鋁、三環己基鋁、三辛基鋁、氫化二乙基鋁、氫化二正丙基鋁、氫化二正丁基鋁、氫化二異丁基鋁(DIBAH)、氫化二己基鋁、氫化二異己基鋁、氫化二辛基鋁、氫化二異辛基鋁、二氫化乙基鋁、二氫化正丙基鋁、二氫化異丁基鋁、氯化二乙基鋁(DEAC)、二氯化單乙基鋁(EADC)、氯化二甲基鋁、氯化二異丁基鋁、二氯化異丁基鋁、倍半氯化乙基鋁(EASC)、以及其中烴取代基中的一個被氫原子取代的相應的化合物以及其中烴取代基中的一個或兩個被異丁基取代的那些。三乙基鋁、三異丁基鋁(TIBA)、氫化二異丁基鋁(DIBAH)是特別地優選的。
優選地,當被用於形成根據本發明的(共)聚合催化體系時,可以以使得在具有通式(I)的鋯的吡啶絡合物中存在的鋯與在具有通式(IV)的烷基鋁中存在的鋁之間的摩爾比可以在從5到5000的範圍內、優選地從10到1000的範圍內的比例,將具有通式(IV)的烷基鋁放置成與具有通式(I)的鋯的吡啶絡合物接觸。具有通式(I)的鋯的吡啶絡合物和具有通式(IV)的烷基鋁被放置成彼此進行接觸的順序不是特別關鍵的。
關於具有通式(IV)的烷基鋁的另外的細節可以在國際專利申請WO 2011/061151中找到。
在特別優選的實施方案中,所述有機氧化化合物(b2)可以選自具有通式(V)的鋁氧烷:
(R9)2-Al-O-[-Al(R10)-O-]p-Al-(R11)2(V)
其中相同或不同的R9、R10和R11代表氫原子、滷素原子比如例如氯、溴、碘、氟;或選自直鏈的或支鏈的C1-C20烷基、環烷基、芳基,所述基團任選地被一個或更多個矽原子或鍺原子取代;並且p是在從0到1000的範圍內的整數。
如已知的,鋁氧烷是通過本領域已知的工藝可獲得的、具有可變的O/Al比率的、包含Al-O-Al鍵的化合物,所述已知的工藝比如例如通過使烷基鋁或烷基鋁滷化物與水或與包含預先確定的量的可用的水的其他化合物在受控條件中反應,比如例如在三甲基鋁與硫酸鋁六水合物、硫酸銅五水合物或硫酸鐵五水合物反應時。
所述鋁氧烷並且特別地甲基鋁氧烷(MAO)是通過有機金屬化學中的已知工藝,比如例如通過將三甲基鋁添加到硫酸鋁水合物在己烷中的懸浮液,可獲得的化合物。
優選地,當被用於形成根據本發明的(共)聚合催化體系時,可以以使得在具有通式(V)的鋁氧烷中存在的鋁(Al)與在具有通式(I)的鋯的吡啶絡合物中存在的鋯之間的摩爾比在從10到10000的範圍內、優選地從100到5000的範圍內的比例,將具有通式(V)的鋁氧烷放置成與具有通式(I)的鋯的吡啶絡合物接觸。具有通式(I)的鋯的吡啶絡合物和具有通式(V)的鋁氧烷被放置成彼此進行接觸的順序不是特別關鍵的。
連同前面提及的優選的具有通式(V)的鋁氧烷,根據本發明的化合物(b2)的定義還包括鎵氧烷(galloxane),其中鎵替代通式(V)中的鋁;和錫氧烷(stannoxane),其中錫替代通式(V)中的鋁,鎵氧烷和錫氧烷作為在金屬茂絡合物的存在下烯烴的聚合助催化劑的用途是已知的。關於所述鎵氧烷和錫氧烷的另外的細節可以例如在美國專利US 5,128,295和US 5,258,475中找到。
對本發明的目的特別有用的具有通式(V)的鋁氧烷的特定的實例是:甲基鋁氧烷(MAO)、乙基鋁氧烷、正丁基鋁氧烷、四異丁基鋁氧烷(TIBAO)、叔丁基鋁氧烷、四(2,4,4-三甲基-戊基)-鋁氧烷(TIOAO)、四(2,3-二甲基-丁基)-鋁氧烷(TDMBAO)、四(2,3,3-三甲基-丁基)-鋁氧烷(TTMBAO)。原樣的甲基鋁氧烷(MAO)或呈乾燥形式的甲基鋁氧烷(MAO-幹)是特別地優選的。
關於具有通式(V)的鋁氧烷的另外的細節可以在國際專利申請WO 2011/061151中找到。
在本發明的優選的實施方案中,所述化合物或化合物的混合物(b3)可以選自鋁的有機化合物和特別地硼的有機化合物,比如例如由以下通式代表的那些:
[(RC)WH4-W]·[B(RD)4]-;B(RD)3;Al(RD)3;B(RD)3P;[Ph3C]+·[B(RD)4]-;[(RC)3PH]+·[B(RD)4]-;
[Li]+·[B(RD)4]-;[Li]+·[Al(RD)4]-
其中w是在從0到3的範圍內的整數,每個RC基團獨立地代表具有從1個到10個碳原子的烷基或芳基,且每個RD基團獨立地代表具有從6個到20個碳原子的被部分地或全部地氟化、優選地被全部地氟化的芳基,P代表任選地被取代的吡咯基。
優選地,當被用於形成根據本發明的(共)聚合催化體系時,可以以使得在化合物或化合物的混合物(b3)中存在的金屬(M')與在具有通式(I)的鋯的吡啶絡合物中存在的鋯(Zr)之間的摩爾比在從0.1到15的範圍內、優選地從0.5到10的範圍內、更優選地從1到6的範圍內的比例,將化合物或化合物的混合物(b3)放置成與具有通式(I)的鋯的吡啶絡合物接觸。具有通式(I)的鋯的吡啶絡合物和化合物或化合物的混合物(b3)被放置成彼此進行接觸的順序不是特別關鍵的。
所述化合物或化合物的混合物(b3)(特別地如果具有通式(I)的鋯的吡啶絡合物中的X1、X2和X3不同於烷基),必須以與具有通式(V)的鋁氧烷比如例如甲基鋁氧烷(MAO)或優選地與具有通式(IV)的烷基鋁、更優選地與在每個烷基殘基中具有從1個到8個碳原子的三烷基鋁比如例如三甲基鋁、三乙基鋁、三異丁基鋁(TIBA)的組合來使用。
如果使用化合物或化合物的混合物(b3),那麼通常被用於提供根據本發明的(共)聚合催化體系的方法的實例在以下的清單中被定性地概述,然而所述清單不以任何方式限制本發明的總體範圍:
(m1)使具有通式(I)的鋯的吡啶絡合物(其中X1、X2和X3中至少一個是烷基)與至少一種化合物或化合物的混合物(b3)(其陽離子能夠與所述烷基反應以形成中性化合物,且其陰離子為體積大的(bulky)、非配位的並且能夠使負電荷離域)接觸;
(m2)使具有通式(I)的鋯的吡啶絡合物與以從10/1到300/1的摩爾過量使用的至少一種具有通式(IV)的烷基鋁、優選地三烷基鋁反應,隨後與以相對於鋯(Zr)的幾乎化學計量的量或稍微過量的強路易斯酸比如三(五氟苯基)硼[化合物(b3)]反應;
(m3)使具有通式(I)的鋯的吡啶絡合物與從10/1到1000/1、優選地從100/1到500/1的摩爾過量的至少一種可以由式AlR」'mZ3-m代表的三烷基鋁或烷基鋁滷化物接觸並且反應,其中R」'是直鏈或支鏈的C1-C8烷基或其混合物,Z是滷素、優選地氯或溴,且m是在從1到3的範圍內的小數(decimal number);隨後向如此獲得的組合物以使得所述化合物或化合物的混合物(b3)或所述化合物或化合物的混合物(b3)的鋁與具有通式(I)的鋯的吡啶絡合物的鋯之間的比率在從0.1到15的範圍內、優選地從1到6的範圍內的量添加至少一種化合物或化合物的混合物(b3)。
能夠用於與根據本發明的具有通式(I)的鋯的吡啶絡合物反應產生離子催化體系的化合物或化合物的混合物(b3)的實例在以下的出版物中被描述,雖然是關於離子金屬茂絡合物的形成,其內容通過引用併入本文:-W.Beck等,"Chemical Reviews"(1988),第88卷,第1405頁-第1421頁;-S.H.Stares,"Chemical Reviews"(1993),第93卷,第927頁-第942頁;-歐洲專利申請EP 277003、EP 495375、EP 520732、EP 427697、EP 421659、EP 418044;
-公布的國際專利申請WO 92/00333、WO 92/05208。
對本發明的目的特別有用的化合物或化合物的混合物(b3)的特定的實例是:三丁基銨-四-五氟苯基-硼酸鹽、三丁基銨-四-五氟苯基-鋁酸鹽、三丁基銨-四-[(3,5-二-(三氟苯基)]-硼酸鹽、三丁基銨-四-(4-氟苯基)]-硼酸鹽、N,N-二甲基苄基銨-四-五氟苯基-硼酸鹽、N,N-二甲基-己基銨-四-五氟苯基-硼酸鹽、N,N-二甲基苯銨-四(五氟苯基)-硼酸鹽、N,N-二甲基苯銨-四(五氟苯基)-鋁酸鹽、二(丙基)銨-四(五氟苯基)-硼酸鹽、二(環己基)銨-四(五氟苯基)硼酸鹽、三苯基碳正離子-四(五氟苯基)-硼酸鹽、三苯基碳正離子-四(五氟苯基)-鋁酸鹽、三(五氟苯基)硼、三(五氟苯基)-鋁、或其混合物。四-五氟苯基-硼酸鹽是優選的。
為了本說明書和以下的權利要求的目的,關於由分子組成的化合物並且關於原子和離子兩者使用術語「摩爾」和「摩爾比」,對於原子和離子,省略術語克原子或原子比(即使這些術語在科學上更正確)。
為了本發明的目的,其他的添加劑或組分可以任選地被添加到前面提及的催化體系以便調節其以在實踐中滿足特定的要求。因此,如此獲得的催化體系應當被認為被包括在本發明的範圍中。可以被添加到根據本發明的催化體系的製劑(preparation)和/或製劑(formulation)的添加劑和/或組分是例如:惰性溶劑,比如例如脂肪族和/或芳香族烴、脂肪族和/或芳香族醚;弱配位的添加劑(例如,路易斯鹼),其選自例如非可聚合的烯烴、空間位阻的或缺電子的醚;滷代劑,比如例如矽滷化物、滷代烴,優選地氯代烴;或其混合物。
所述催化體系如先前陳述的,可以通過本領域已知的方法來製備。
例如,所述催化體系可以被單獨地製備(預形成)並且隨後被引入到(共)聚合環境中。為了此目的,所述催化體系可以通過使至少一種具有通式(I)的鋯的吡啶絡合物(a)與至少一種助催化劑(b)、任選地在選自上文列出的那些的其他的添加劑或組分的存在下、在溶劑比如例如甲苯、庚烷的存在下、在從20℃到60℃的範圍內的溫度下反應持續在從10秒到10小時的範圍內、優選地從30秒到5小時的範圍內的時間來製備。關於製備所述催化體系的更多的細節可以在下文報告的實施例中找到。
可選擇地,所述催化體系可以在原位(in situ)即直接在(共)聚合環境中製備。為了此目的,所述催化體系可以通過分別引入具有通式(I)的鋯的吡啶絡合物(a)、助催化劑(b)和待被(共)聚合的預選的共軛二烯,在進行(共)聚合的條件下操作來製備。
為了本發明的目的,前面提及的催化體系還可以被負載在優選地由矽氧化物和/或鋁氧化物比如例如二氧化矽、氧化鋁或矽鋁酸鹽形成的惰性固體上。已知的負載方法可以被用於負載所述催化體系,所述負載方法通常包括在合適的惰性液體介質中在任選地通過加熱到高於200℃的溫度活化的載體與催化體系即本發明的目標的組分(a)和(b)中的一種或兩種之間的接觸。為了本發明的目的,對於組分中的兩種都被負載是不必要的,也可能的是對於僅具有通式(I)的鋯的吡啶絡合物(a)或助催化劑(b)在載體的表面上存在。在後者的情況下,當期望形成用於聚合的活性催化劑的時刻,表面中不存在的組分隨後被放置成與負載的組分接觸。
已經通過使所述固體官能化並且形成在固體和具有通式(I)的鋯的吡啶絡合物之間的共價鍵而被負載在固體上的具有通式(I)的鋯的吡啶絡合物和基於其的催化體系,也被包括在本發明的範圍中。
另外,本發明涉及使共軛二烯(共)聚合的工藝,其特徵是使用所述催化體系。
可以被用於使共軛二烯(共)聚合的具有通式(I)的鋯的吡啶絡合物(a)和助催化劑(b)的量取決於期望實施的(共)聚合工藝而變化。然而,所述量使得獲得在上文陳述的值的範圍內的在具有通式(I)的鋯的吡啶絡合物中存在的鋯(Zr)與在助催化劑(b)中存在的金屬(例如,鋁,如果助催化劑(b)選自烷基鋁(b1)或鋁氧烷(b2);硼,如果助催化劑(b)選自具有通式(III)的化合物或化合物的混合物(b3))之間的摩爾比。
可以使用根據本發明的催化體系(共)聚合的共軛二烯的特定的實例是:1,3-丁二烯、2-甲基-1,3-丁二烯(異戊二烯)、2,3-二甲基-1,3-丁二烯、1,3-戊二烯、1,3-己二烯、環-1,3-己二烯(cyclo-1,3-hexadiene)。1,3-丁二烯是優選的。前面提及的可(共)聚合的共軛二烯可以單獨使用,或以兩種或更多種二烯的混合物使用。在後者的情況下.當使用兩種或更多種二烯的混合物時,將獲得共聚物。
在特別優選的實施方案中,本發明涉及用於使1,3-丁二烯聚合的工藝,其特徵是使用所述催化體系。
通常來說,所述(共)聚合可以在聚合溶劑(polymerization solvent)的存在下進行,所述聚合溶劑通常選自惰性有機溶劑比如例如:飽和的脂肪族烴,比如例如丁烷、戊烷、己烷、庚烷或其混合物;飽和的脂環族烴,比如例如環戊烷、環己烷、環己烷或其混合物;單烯烴,比如例如1-丁烯、2-丁烯或其混合物;芳香族烴,比如例如苯、甲苯、二甲苯或其混合物;滷代烴,比如例如二氯甲烷、氯仿、四氯化碳、三氯乙烯、全氯乙烯、1,2-二氯乙烷、氯苯、溴苯、氯甲苯或其混合物。優選地,(共)聚合溶劑選自飽和的脂肪族烴。
可選擇地,所述(共)聚合可以使用與(共)聚合溶劑相同的待被(共)聚合的共軛二烯在被稱為本體工藝(bulk process)的工藝中來進行。
通常來說,待被(共)聚合的共軛二烯在所述(共)聚合溶劑中的濃度在相對於共軛二烯和惰性有機溶劑的混合物的總重量的從按重量計5%到按重量計50%的範圍內、優選地從按重量計10%到按重量計20%的範圍內。
通常來說,所述(共)聚合工藝可以在從-70℃到+100℃的範圍內、優選地從-20℃到+80℃的範圍內的溫度下進行。
就壓力而言,在待被(共)聚合的混合物的組分的壓力下操作是優選的。
所述(共)聚合可以連續地或分批地進行。
如上文所陳述的,所述工藝使得獲得具有高的1,4-反式單元含量,即,1,4-反式單元含量≥94%的共軛二烯的(共)聚合物例如直鏈或支鏈的聚丁二烯是可能的。
為了更好地理解本發明的目的並且為了將其實施,在下文給出某些說明性、非限制性實例。
實施例
試劑和材料
以下清單陳述在本發明的以下實施例中使用的試劑和材料、其任何任選的預處理和其製造商:
-2,6-二異丙基苯胺(Aldrich):按原樣使用;
-2-叔丁基苯胺(Aldrich):按原樣使用;
-2,6-苯甲醯基吡啶(Aldrich):按原樣使用;
-苯胺(Aldrich):在減壓下蒸餾並且保持在惰性氣氛中;
-2,4,6-三甲基苯胺(Aldrich):按原樣使用;
-2-吡啶甲醛(Aldrich):按原樣使用;
-2-乙醯基吡啶(Aldrich):按原樣使用;
-二氯甲烷(Carlo Erba,RPE):按原樣使用;
-甲醇(Carlo Erba,RPE):按原樣使用,或任選地通過經鎂(Mg)蒸餾而被脫水(anhydrify);
-硼氫化鈉(Aldrich):按原樣使用;
-乙酸乙酯(Aldrich):按原樣使用;
-己烷(Aldrich):純的,≥99%,在惰性氣氛下經鈉(Na)蒸餾;
-乙醚(Aldrich):按原樣使用;
-甲酸(Aldrich):按原樣使用;
-庚烷(Aldrich):純的,≥99%,在惰性氣氛下經鈉(Na)蒸餾;
-硫酸鈉(Aldrich):按原樣使用;
-氯仿(Aldrich):按原樣使用;
-甲苯(Aldrich):純的,≥99.5%,在惰性氣氛下經鈉(Na)蒸餾;
-四氯化鋯(ZrCl4)(Stream Chemicals):按原樣使用;
-四氯化鋯:四氫呋喃絡合物(1:2)[ZrCl4(THF)2](Aldrich):按原樣使用;
-四氫呋喃(THF)(Carlo Erba,RPE):經鉀/二苯甲酮被保持在回流下,且然後在氮氣下蒸餾;
-正丁基鋰(Aldrich):按原樣使用;
-1,3-丁二烯(Air Liquide):純的,≥99,5%,在任何生產前從容器中蒸發,通過經過用分子篩填塞的柱乾燥並且在已被預冷卻到-20℃的反應器之內冷凝;
-甲基鋁氧烷(MAO)(在按重量計10%下的甲苯溶液)(Aldrich):按原樣使用,或呈通過在真空下除去游離的三甲基鋁連同來自甲苯溶液的溶劑並且仍舊在真空下將獲得的殘餘物乾燥獲得的乾燥形式(MAO-幹);
-以37%的在水溶液中的鹽酸(Aldrich):按原樣使用。
-三異丁基鋁(TIBA)(Aldrich):按原樣使用;
-氘代四氯乙烯(C2D2Cl4)(Acros):按原樣使用;
-氘代氯仿(CDCl3)(Acros):按原樣使用。
使用以下分析和表徵方法。
元素分析
a)鋯的確定
為了確定在被用於本發明的目的的鋯的吡啶絡合物中按重量計鋯(Zr)的量,在氮氣流下在乾燥箱中操作下,將精確地稱重的約30mg-50mg的樣品的等分部分連同1ml的40%氫氟酸(HF)、0.25ml的96%硫酸(H2SO4)和1ml的70%硝酸(HNO3)的混合物放置於約30ml的鉑坩堝中。隨後,坩堝在板上被加熱,增加溫度直到出現白色硫磺煙霧(約200℃)。如此獲得的混合物被冷卻到室溫(20℃-25℃),1ml的70%硝酸(HNO3)被添加且使其回到煙霧出現的點(point)。在重複該序列另外兩次之後,獲得澄清的且幾乎無色的溶液。隨後,在冷卻時,添加1ml的硝酸(HNO3)和約15ml的水,同時加熱到80℃持續約30分鐘。將如此製備的樣品用Milli-Q-純度的水稀釋直到精確地稱重的約50g的重量,以獲得溶液,使用Thermo Optek IRIS Advantage Duo ICP-OES(光學發射等離子體(optical emission plasma))分光計,通過與已知濃度的溶液比較,對所述溶液進行儀器分析測定。為了此目的,對於每種分析物,在0ppm-10ppm的範圍內,通過測量通過按重量稀釋認證的溶液(certified solution)獲得的已知滴定度的溶液來製備校準曲線。
在進行分光光度檢測之前,如上文製備的樣品的溶液被進一步按重量稀釋以便獲得接近參考濃度的濃度。所有樣品被一式兩份地製備。如果一式兩份測試的單獨的數據相對於其平均值相差不超過2%,那麼結果被認為是可接受的。
b)氯的確定
為了此目的,在乾燥箱中在氮氣流下,將約30mg-50mg的被用於本發明的目的的鋯的吡啶絡合物的樣品在100ml的玻璃燒杯中精確地稱重。添加2g的碳酸鈉(Na2CO3)並且在乾燥箱之外添加50ml的Mili-Q水。在板上在磁力攪拌下使此達到沸騰持續約30分鐘。將其留下冷卻,添加1/5稀硫酸(H2SO4)直到酸性反應,並且用電位滴定儀、使用硝酸銀(AgNO3)0.1N進行滴定。
c)碳、氫和氮的確定
使用自動Carlo Erba 1106分析儀,確定在被用於本發明的目的的鋯的吡啶絡合物以及被用於本發明的目的的配體中的碳、氫和氮。
13C-HMR和1H-HMR光譜
使用Bruker Avance 400核磁共振分光計,在103℃下使用氘代四氯乙烯(C2D2Cl4)和作為內標的六甲基二矽氧烷(HDMS),或在25℃下使用氘代氯仿(CDCl3)和作為內標的四甲基矽烷(TMS)記錄13C-HMR和1H-HMR光譜。為了此目的,使用具有相對於聚合物溶液的總重的按重量計10%的濃度的聚合物溶液。
通過基於在由Mochel,V.D,在「Journal of Polymer Science Part A-1:Polymer Chemistry」(1972),第10卷,第4期,第1009頁-第1018頁的文獻中報告的分析前面提及的光譜,確定聚合物的微結構[即,1,4-反式單元含量(%)]。
FTIR-ATR光譜
使用裝配有Thermo Spectra-Tech水平ATR接頭(connection)的Bruker IFS 48分光光度計記錄FTIR-ATR光譜。其中待被分析的樣品所放置於的部分是Fresnel ATR配件(Shelton,CT,USA),所述Fresnel ATR配件使用在水平方向上以45°的入射角的硒化鋯晶體(ZnSe)。
在本發明中使用的鋯的吡啶絡合物的FTIR-TR光譜通過將待被分析的鋯的吡啶絡合物的樣品插入到所述部分中來獲得。
FT-IR光譜
FT-IR光譜使用Thermo Nicolet Nexus 670和Bruker IFS 48分光光度計來記錄。
聚合物的FT-IR光譜從溴化鉀(KBr)片上的聚合物膜獲得,所述膜通過沉積待被分析的聚合物在熱的鄰二氯苯中的溶液獲得。分析的聚合物溶液的濃度是相對於聚合物溶液的總重量的按重量計10%。
熱分析(DSC)
為了確定獲得的聚合物的熔點(Tm)和結晶溫度(Tc)的目的,使用Perkin Elmer Pyris差示掃描量熱計進行DSC(「差示掃描量熱法」)熱分析。為了此目的,在惰性氮氣氣氛下,以在從1℃/min到20℃/min的範圍內的掃描速率分析5mg的聚合物。
分子量的測定
獲得的聚合物的分子量(MW)通過GPC(「凝膠滲透色譜法」)在以下條件下操作進行:
-Agilent 1100泵;
-Agilent 1100I.R.檢測器;
-Mixed-A PL柱;
-溶劑/洗脫劑:四氫呋喃(THF);
-流速:1ml/min;
-溫度:25℃
-分子量的計算:通用校準方法(Universal Calibration method)。
給出重均分子量(Mw)和對應於比率Mw/Mn(Mn=數均分子量)的「多分散性指數」(PDI)。
氣相色譜-質譜法(GC-MS)
使用Thermo ISQ單-四級杆質譜分光計進行氣相色譜-質譜法(GC-MS)。為了此目的,使待被分析的配體的樣品在二氯甲烷(CH2Cl2)中以0.1mg/ml的濃度溶解,並且使用在以下條件下操作的所述分光計分析:
-離子化手段:電子電離(EI);
-GC升溫速率(GC ramp):50℃每2min;以10℃/min的速率加熱直到300℃;
-注射器溫度:300℃;
-注射體積:1.30μl;
-「轉移線」溫度(「transfer line」temperature):280℃;
-離子源溫度250℃;
-四級杆掃描參數:在0.2s的掃描時間下的35amu–500amu。
實施例1
具有式(L1)的配體的合成
1.1具有式(L1a)的化合物的合成
在設置有用於共沸水除去的迪安-斯達克分水器的500ml燒瓶中,向2,6-二異丙基苯胺(27.93g,157.5mmol)在二氯甲烷(300ml)中的溶液添加2-吡啶甲醛(16.86g,157.5mmol)。獲得的混合物在回流下被加熱持續20小時,並且隨後在真空下乾燥,以獲得對應於具有式(L1a)的化合物的41.7g的黃色固體(收率=99%)。
元素分析[實測的(計算的)]:C:81.14%(81.16%);H:8.33%(8.32%);N:10.6%(10.52%)。
1H-NMR(CDCl3,δppm):8.72(d,1H,PyH),8.32(s,1H CH=N),8.27(d,1H PyH),7.86(t,1H PyH),7.39(m,1H PyH),7.11-7.20(m,3H ArH),3.00(sept,2H CHMe2),1.18(d,12H C(CH3)2)。
1.2具有式(L1)的配體的合成
向設置有攪拌器的2升反應器中,裝載28g(105.1mmol)的如上文描述獲得的具有式(L1a)的化合物和1800ml的無水甲醇:使整體冷卻到0℃並且隨後以小份的方式添加硼氫化鈉(70g,1850mmol)。將獲得的混合物留在室溫下在攪拌下過夜,並且隨後用鹽水猝滅並且使用乙酸乙酯萃取。隨後,溶劑通過在減壓下蒸餾來除去,並且獲得的殘餘物通過在矽膠色譜柱中洗脫[洗脫劑:以9/1比率(v/v)的己烷/乙酸乙酯混合物]來純化,並且隨後用冷的乙醚處理,以獲得對應於具有式(L1)的配體的16.9g的白色結晶固體(收率=60%)。
GC-MS:M+=m/z 268;[M-C3H7]+=m/z 225;[M-C6H6N]+=m/z 176;m/z 93C6H7N。
1H-NMR(CDCl3,δppm):8.61(d,1H,o-PyH),7.66(td,1H,PyH),7.30(d,1H,PyH),7.21(m,1H,PyH),7.04-7.12(m,3H,ArH),4.20(s,2H,CH2),4.10(s,1H,NH),3.47(m,2H,-CH(CH3)2),1.42(d,12H,-CH(CH3)2)。
實施例2
具有式(L2)的配體的合成
2.1具有式(L2a)的化合物的合成
在500ml燒瓶中,向2,6-二異丙基苯胺(13.3g,75mmol)在甲醇(300ml)中的溶液添加2-乙醯基吡啶(9.1g,75mmol):將獲得的混合物留在室溫下在攪拌下持續48小時。將獲得的沉澱物過濾,並且隨後在真空下乾燥,以獲得對應於具有式(L2a)的化合物的14g的黃色結晶粉末(收率=67%)。
元素分析[實測的(計算的)]:C:81.37%(81.38%);H:8.64%(8.63%);N:10.01%(9.99%)。
1H-NMR(CDCl3,δppm)8.69(d,1H,PyH),8.38(d,1H,PyH),7.82(t,1H,PyH),7.39(m,1H,PyH),7.11-7.20(m,3H,ArH),2.75(m,2H,CHMe2),2.21(s,3H,N=CH-Me),1.15(d,12H,CH(CH3)2)。
2.2具有式(L2)的配體的合成
向設置有攪拌器的2升反應器中,裝載24g(85mmol)的如上文描述獲得的具有式(L2a)的化合物和900ml的無水甲醇:使整體冷卻到0℃並且隨後以小份的方式添加硼氫化鈉(48.6g,1285mmol)。將獲得的混合物留在室溫下在攪拌下過夜,並且隨後用鹽水猝滅並且使用乙酸乙酯萃取。隨後,溶劑通過在減壓下蒸餾來除去,並且獲得的殘餘物通過在矽膠色譜柱中洗脫[洗脫劑:以9/1比率(v/v)的己烷/乙酸乙酯混合物]來純化,並且隨後用冷的乙醚處理,以獲得對應於具有式(L2)的配體的11g的白色結晶固體(收率=46%)。
元素分析[實測的(計算的)]:C:81.03%(80.80%);H:9.42%(9.28%);N:10.01%(9.92%)。
GC-MS:M+=m/z 282;[M-C3H7]+=m/z 239;[M-C7H8N]+=m/z 176;[M-C12H18N]+=m/z 106。
1H-NMR(CDCl3,δppm):8.64(d,1H,HPy),7.53(dt,1H,HPy),7.2(d,1H,HPy),7.00-7.12(m,1H,HPy;m,3H,ArH),4,0-4,2(m,1H,NCH(CH3);m,1H,NH),3.30(sept,2H,-CH(CH3)2),1.55(d,3H,-NCH(CH3)),1.10(s,12H,-CH(CH3)2)。
實施例3
具有式(L3)的配體的合成
3.1具有式(L3a)的化合物的合成
在500ml燒瓶中,向2-叔丁基苯胺(15.89g,106.5mmol)在甲醇(300ml)中的溶液添加2-乙醯基吡啶(12.9g,106.5mmol):將獲得的混合物留在室溫下在攪拌下持續48小時。隨後,溶劑通過蒸發除去並且使用甲醇使獲得的殘餘物結晶,以獲得對應於具有式(L3a)的化合物的20g的黃色結晶粉末(收率=75%)。
元素分析[實測的(計算的)]:C:81.17%(80.91%);H:8.14%(7.99%);N:10.91%(11.10%)。
3.2具有式(L3)的配體的合成
向設置有攪拌器的2升反應器中,裝載28g(111mmol)的如上文描述獲得的具有式(L3a)的化合物和800ml的無水甲醇:使整體冷卻到0℃並且隨後以小份的方式添加硼氫化鈉(38g,1004mmol)。將獲得的混合物留在室溫下在攪拌下過夜,並且隨後用鹽水猝滅並且使用乙酸乙酯萃取。隨後,溶劑通過在減壓下蒸餾來除去,並且獲得的殘餘物通過在矽膠色譜柱中洗脫[洗脫劑:以9/1比率(v/v)的己烷/乙酸乙酯混合物]來純化,並且隨後用冷的乙醚處理,以獲得對應於具有式(L3)的配體的11g的白色結晶固體(收率=39%)。
元素分析[實測的(計算的)]:C:80.00%(80.27%);H:9.12%(8.72%);N:11.31%(11.01%)。
GC-MS:M+=m/z 254;[M-CH3]+=m/z 239;[M-C4H9]+=m/z 197;m/z=183;m/z 132C7H10N2;[M-C10H14N]+=m/z 106;[M-C12H18N]+=m/z 78。
1H-NMR(CDCl3,δppm):8.64(d,1H,HPy),7.7(td,1H,PyH),7.36(d,1H,HPy),7.25(d,1H,ArH),7.18(td,1H,PyH),6.98(td,1H,PyH),6.98(td,1H,PyH),6.48(d,1H,PyH),5.0(寬的單峰,1H,NH),4.7(q,1H,NCH(CH3)),1.57(d,3H,-NCH(CH3)),1.5(s,9H,-C(CH3)3)。
實施例4
具有式(L4)的配體的合成
4.1具有式(L4a)的化合物的合成
在500ml燒瓶中,向2-苯甲醯基吡啶(20g,109mmol)在甲醇(200ml)中的溶液添加苯胺(11.2g,120mmol)和若干滴甲酸:將獲得的混合物留在室溫下在攪拌下持續48小時。隨後,獲得的混合物在減壓下乾燥,並且獲得的殘餘物通過在矽膠色譜柱中洗脫[洗脫劑:以99/1比率(v/v)的庚烷/乙酸乙酯混合物]來純化,以獲得對應於具有式(L4a)的化合物的14.4g的黃色的油(收率=51%)。
元素分析[實測的(計算的)]:C:84.00%(83.69%);H:5.83%(5.46%);N:11.52%(10.84%)。
GC-MS:M+=m/z 258;m/z 180,155,77,51。
4.2具有式(L4)的配體的合成
向設置有攪拌器的2升反應器中,裝載14g(85mmol)的如上文描述獲得的具有式(L4a)的化合物和900ml的無水甲醇:使整體冷卻到0℃並且隨後以小份的方式添加硼氫化鈉(31g,819mmol)。將獲得的混合物留在室溫下在攪拌下過夜,並且隨後用鹽水猝滅並且使用乙酸乙酯萃取。隨後,溶劑通過在減壓下蒸餾來除去,並且獲得的殘餘物通過在矽膠色譜柱中洗脫[洗脫劑:以9/1比率(v/v)的己烷/乙酸乙酯混合物]來純化,並且隨後用冷的乙醚處理,以獲得對應於具有式(L4)的配體的12.5g的白色結晶固體(收率=56.5%)。
元素分析[實測的(計算的)]:C:83.30%(83.04%);H:6.87%(6.19%);N:11.01%(10.76%)。
GC-MS:M+=m/z 260;m/z 182,168,104,77 51。
1H-NMR(CDCl3,δppm):8.6(m 1H,PyH),7.62-7.69(m 1H,PyH),7.45-7.50(m 2H,ArH),7.30-7.38(m,1H,HPy;m 2H,ArH),7.23-7.27(m,1H,ArH),7.18-7.21(m,1H,PyH),7.05-7.13(m,2H,NH-ArH),6.60-6.65(m,3H,NH-ArH),5.55(s,1H,NH),5.50(s,1H,-NCH)。
實施例5
具有式(L5)的配體的合成
5.1具有式(L5a)的化合物的合成
在500ml燒瓶中,向苯胺(26.1g,280mmol)在甲醇(250ml)中的溶液添加2-吡啶甲醛(30g,280mmol)和若干滴甲酸:將獲得的混合物留在室溫下在攪拌下持續48小時。隨後,獲得的混合物通過在減壓下乾燥,並且獲得的殘餘物通過在矽膠色譜柱中洗脫[洗脫劑:以99/1比率(v/v)的庚烷/乙酸乙酯混合物]來純化,以獲得對應於具有式(L5a)的化合物的38g的黃色固體(收率=74.5%)。
元素分析[實測的(計算的)]:C:80.00%(79.10%);H:5.83%(5.53%);N:15.71%(15.37%)。
1H-NMR(CDCl3,δppm)8.70(d,1H,HPy),8.59(s,1H CH=N),8.19(d,1H,HPy),7.77(dt,1H,HPy),7.23-7.42(m,1H,HPy;m,5H,Ar)。
5.2具有式(L5)的配體的合成
向設置有攪拌器的2升反應器中,裝載13g(71.3mmol)的如上文描述獲得的具有式(L5a)的化合物和700ml的無水甲醇:使整體冷卻到0℃並且隨後以小份的方式添加硼氫化鈉(40g,1057mmol)。將獲得的混合物留在室溫下在攪拌下過夜,並且隨後用鹽水猝滅並且使用乙酸乙酯萃取。隨後,溶劑通過在減壓下蒸餾來除去,並且獲得的殘餘物通過在矽膠色譜柱中洗脫[洗脫劑:以9/1比率(v/v)的己烷/乙酸乙酯混合物]來純化,並且隨後用冷的乙醚處理,以獲得對應於具有式(L5)的配體的9.12g的白色結晶固體(收率=69.5%)。
GC-MS:M+=m/z 184;[M-C6H6N]+=m/z 106;[M-C7H7N2]+=m/z 77。
1H-NMR(CDCl3,δppm):8.60(dd,1H,PyH),7.64(m,1H,PyH),7.35(d,1H,PyH),7.22-7.17(m,1H,Py,2H,ArH),6.75(dt,1H,ArH),6.69(d,2H,ArH),4.8(s,1H,NH),4.48(s,2H,Py-CH2N)。
實施例6
具有式(L6)的配體的合成
6.1具有式(L6a)的化合物的合成
在500ml燒瓶中,向2,6-二甲基苯胺(31g,250mmol)在甲醇(250ml)中的溶液添加2-吡啶甲醛(26.8g,250mmol)和若干滴甲酸:將獲得的混合物留在室溫下在攪拌下持續24小時。隨後,將獲得的混合物經硫酸鈉乾燥並且過濾,並且將溶劑通過在真空下的蒸發除去:將獲得的殘餘物用冷的甲醇洗滌,以獲得對應於具有式(L6a)的化合物的47g的橙色固體(收率=89%)。
元素分析[實測的(計算的)]:C:80.00%(79.97%);H:6.81%(6.71%);N:13.71%(13.37%)。
1H-NMR(CDCl3,δppm)8.70(d,1H,HPy),8.33(s,1H,CH=N),8.23(d,1H,HPy),7.82(dt,1H,HPy),7.38(ddd,1H,HPy),6.91-7.15(m,5H,Ar),2.16(s,6H,Ar-CH3)。
6.2具有式(L6)的配體的合成
向設置有攪拌器的2升反應器中,裝載18g(85.6mmol)的如上文描述獲得的具有式(L6a)的化合物和800ml的無水甲醇:使整體冷卻到0℃並且隨後以小份的方式添加硼氫化鈉(24g,634mmol)。將獲得的混合物留在室溫下在攪拌下過夜,並且隨後用鹽水猝滅並且使用乙酸乙酯萃取。隨後,溶劑通過在減壓下蒸餾來除去,並且獲得的殘餘物通過在矽膠色譜柱中洗脫[洗脫劑:以9/1比率(v/v)的己烷/乙酸乙酯混合物]來純化,並且隨後用冷的乙醚處理,以獲得對應於具有式(L6)的配體的9.15g的白色結晶固體(收率=50.4%)。
GC-MS:M+=m/z 212;[M-C6H6N]+=m/z 120。
1H-NMR(CDCl3,δppm):8.63(d,1H,PyH),7.65(dt,1H,PyH),7.27(d,1H,PyH),7.20(dd,1H,PyH),7.02(d,2H,ArH),6.85(m,1H,ArH),4.4(寬的單峰,1H,NH),4.31(s,2H,Py-CH2N),2.35(s,6H,ArCH3)。
實施例7
具有式(L7)的配體的合成
7.1具有式(L7a)的化合物的合成
在500ml燒瓶中,向2,4,6-三甲基苯胺(12.6g,93mmol)在甲醇(250ml)中的溶液添加2-吡啶甲醛(10g,93mmol)和若干滴甲酸:將獲得的混合物留在室溫下在攪拌下持續48小時。隨後,溶劑通過在真空下的蒸發除去,並且獲得的油狀殘餘物通過在矽膠色譜柱中洗脫[洗脫劑:以99/1比率(v/v)的庚烷/乙酸乙酯混合物]來純化,以獲得對應於具有式(L7a)的化合物的17g的淡黃色固體(收率=81%)。
元素分析[實測的(計算的)]:C:80.56%(80.32%);H:7.22%(7.19%);N:13.11%(12.49%)。
GC-MS:M+=m/z 224;[M-CH3]+=m/z 209;[M-C5H4N]+=m/z 146。
1H-NMR(CDCl3,δppm)8.70(m,1H,HPy),8.35(s,1H CH=N),8.29(d,1H,HPy),7.84(tdd,1H,HPy),7.41(m,1H,HPy),6.91(s,2H ArH),2.31(s,3H Ar(CH3)),2.10(s,6H Ar(CH3)2)。
7.2具有式(L7)的配體的合成
向設置有攪拌器的2升反應器中,裝載13g(58mmol)的如上文描述獲得的具有式(L7a)的化合物、80ml的無水甲醇、80ml的氯仿、和以小份方式的硼氫化鈉(2.2g,58mmol)。將獲得的混合物留在室溫下在攪拌下過夜。隨後,將溶劑通過在減壓下的蒸餾除去,並且使用乙酸乙酯(50ml)和水(50ml)的混合物萃取獲得的殘餘物。使用水洗滌獲得的有機萃取物直到中性,經硫酸鈉脫水,過濾,並且經歷在減壓下的蒸餾以除去剩餘的溶劑,以獲得油狀黃色殘餘物。向所述油狀殘餘物添加25ml的冷的庚烷,以獲得對應於具有式(L7)的配體的5.15g的白色結晶固體(收率=39%)。
GC-MS:M+=m/z 226;[M-C6H6N]+=m/z 134。
1H-NMR(CDCl3,δppm):8.59(d,1H,PyH),7.65(dt,1H,PyH),7.27(d,1H,PyH),7.20(m,1H,PyH),6.8(d,2H,ArH),4.2(s,2H,Py-CH2N),4.1(寬的單峰,1H,NH),2.28(s,6H,ArCH3),2.2(s,3H,Ar-CH3)。
實施例8
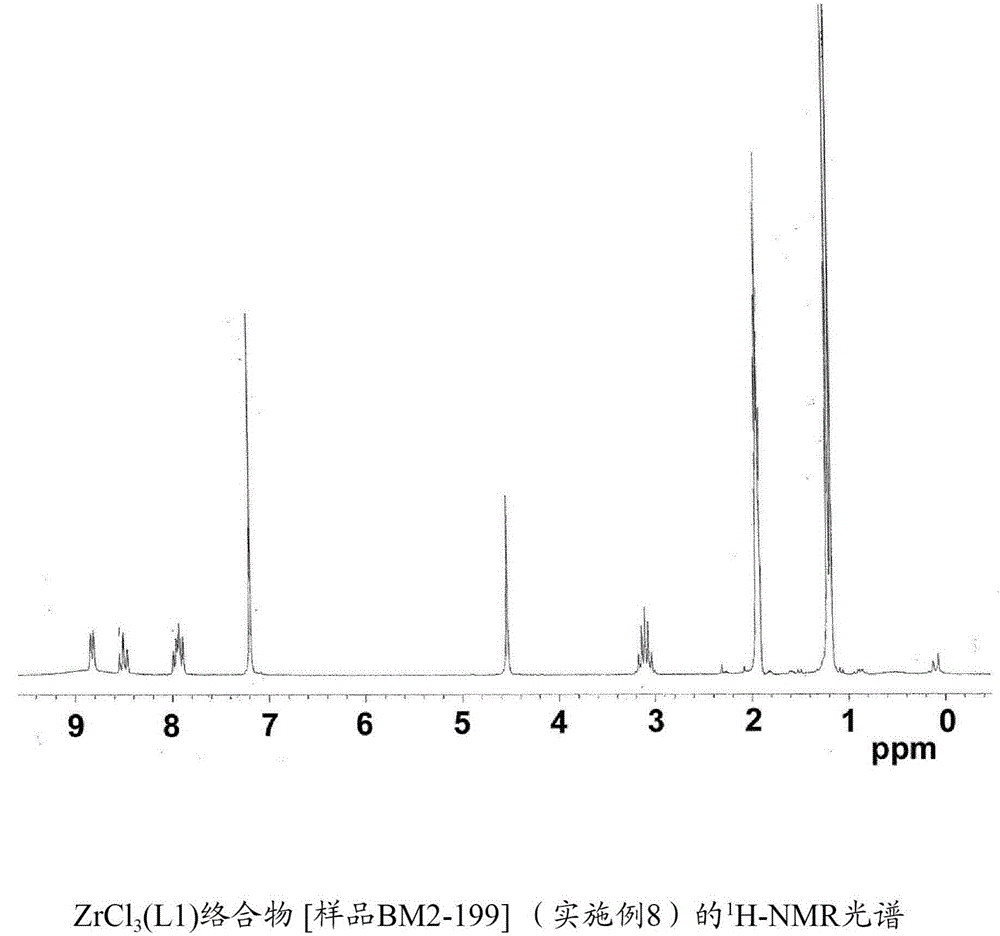
ZrCl3(L1)[樣品BM2-199]的合成
將四氯化鋯(ZrCl4)(0.500g;2.14mmol)連同如在實施例1中描述獲得的具有式(L1)的配體(0.599g;2.22mmol;L1/Zr摩爾比=1.03)在甲苯(15ml)中的溶液引入到100ml的長頸燒瓶中。將獲得的混合物留在室溫下在攪拌下持續30分鐘,並且隨後在回流下加熱持續2小時。將形成固體通過過濾回收,用庚烷(2×2ml)洗滌並且在減壓下在室溫下乾燥,以獲得對應於ZrCl3(L1)絡合物的0.66g(收率=66%)的澄清的黃色微晶固體產物。
元素分析[實測的(計算的)]:C:45.87%(46.49%);H:4.65%(4.98%);N:5.45%(6.02%);Zr:18.72%(19.62%);Cl:21.65%(22.87%)。
圖1示出獲得的ZrCl3(L1)絡合物的FTIR-ATR光譜。
圖2示出獲得的ZrCl3(L1)絡合物的1H-NMR光譜。
實施例9
ZrCl3(L2)[樣品BM2-207]的合成
將四氯化鋯(ZrCl4)(0.398g;1.71mmol)連同如在實施例2中描述獲得的具有式(L2)的配體(0.507g;1.80mmol;L2/Zr摩爾比=1.05)在甲苯(10ml)中的溶液引入到100ml的長頸燒瓶中。將獲得的混合物留在室溫下在攪拌下持續30分鐘,並且隨後在回流下加熱持續2小時。將形成的固體通過過濾回收,用庚烷(2×2ml)洗滌並且在減壓下在室溫下乾燥,以獲得對應於ZrCl3(L2)絡合物的0.71g(收率=86%)的澄清的黃色微晶固體產物。
元素分析[實測的(計算的)]:C:46.87%(47.64%);H:4.85%(5.26%);N:5.21%(5.84%);Zr:19.87%(19.04%);Cl:21.89%(22.20%)。
圖3示出獲得的ZrCl3(L2)絡合物的FTIR-ATR光譜。
圖4示出獲得的ZrCl3(L2)絡合物的1H-NMR光譜。
實施例10
ZrCl3(L3)[樣品MT-2]的合成
將四氯化鋯(ZrCl4)(0.525g;2.25mmol)連同如在實施例3中描述獲得的具有式(L3)的配體(0.570g;2.24mmol;L3/Zr摩爾比=1)在甲苯(10ml)中的溶液引入到100ml的長頸燒瓶中。將獲得的混合物留在室溫下在攪拌下持續30分鐘,並且隨後在回流下加熱持續2小時。將形成的固體通過過濾回收,用庚烷(2×2ml)洗滌並且在減壓下在室溫下乾燥,以獲得對應於ZrCl3(L3)絡合物的0.81g(收率=80%)的澄清的黃色微晶固體產物。
元素分析[實測的(計算的)]:C:44.82%(45.28%);H:4.05%(4.69%);N:5.95%(6.21%);Zr:19.99%(20.23%);Cl:23.00%(23.58%)。
實施例11
ZrCl3(L5)[樣品MT-4]的合成
將四氯化鋯(ZrCl4)(0.368g;1.58mmol)連同如在實施例5中描述獲得的具有式(L5)的配體(0.289g;1.58mmol;L5/Zr摩爾比=1)在甲苯(10ml)中的溶液引入到100ml的長頸燒瓶中。將獲得的混合物留在室溫下在攪拌下持續30分鐘,並且隨後在回流下加熱持續2小時。將形成的固體通過過濾回收,用庚烷(2×2ml)洗滌並且在減壓下在室溫下乾燥,以獲得對應於ZrCl3(L5)絡合物的0.26g(收率=43%)的澄清的黃色微晶固體產物。
元素分析[實測的(計算的)]:C:36.87%(37.85%);H:2.65%(2.91%);N:6.95%(7.36%);Zr:22.98%(23.95%);Cl:27.42%(27.93%)。
圖5示出獲得的ZrCl3(L5)絡合物的FTIR-ATR光譜。
實施例12
ZrCl3(L6)[樣品MT-30]的合成
將四氯化鋯(ZrCl4)(0.317g;1.36mmol)連同如在實施例6中描述獲得的具有式(L6)的配體(0.289g;1.36mmol;L6/Zr摩爾比=1)在甲苯(10ml)中的溶液引入到100ml的長頸燒瓶中。將獲得的混合物留在室溫下在攪拌下持續30分鐘,並且隨後在回流下加熱持續2小時。將形成的固體通過過濾回收,用庚烷(2×2ml)洗滌並且在減壓下在室溫下乾燥,以獲得對應於ZrCl3(L6)絡合物的0.50g(收率=90%)的澄清的黃色微晶固體產物。
元素分析[實測的(計算的)]:C:41.52%(41.12%);H:3.15%(3.70%);N:6.15%(6.85%);Zr:21.95%(22.31%);Cl:25.75%(26.01%)。
實施例13
ZrCl3(L7)[樣品MT-52]的合成
將四氯化鋯(ZrCl4)(0.351g;1.51mmol)連同如在實施例7中描述獲得的具有式(L7)的配體(0.341g;1.51mmol;L7/Zr摩爾比=1)在甲苯(10ml)中的溶液引入到100ml的長頸燒瓶中。將獲得的混合物留在室溫下在攪拌下持續30分鐘,並且隨後在回流下加熱持續2小時。將形成的固體通過過濾回收,用庚烷(2×2ml)洗滌並且在減壓下在室溫下乾燥,以獲得對應於ZrCl3(L7)絡合物的0.50g(收率=78%)的澄清的黃色微晶固體產物。
元素分析[實測的(計算的)]:C:42.00%(42.60%);H:3.75%(4.05%);N:6.01%(6.62%);Zr:20.87%(21.57%);Cl:24.98%(25.15%)。
實施例14
ZrCl3(L4)[樣品MT-56]的合成
將四氯化鋯(ZrCl4)(0.212g;0.910mmol)連同如在實施例4中描述獲得的具有式(L4)的配體(0.236g;0.910mmol;L4/Zr摩爾比=1)在甲苯(10ml)中的溶液引入到100ml的長頸燒瓶中。將獲得的混合物留在室溫下在攪拌下持續30分鐘,並且隨後在回流下加熱持續2小時。將形成的固體通過過濾回收,用庚烷(2×2ml)洗滌並且在減壓下在室溫下乾燥,以獲得對應於ZrCl3(L4)絡合物的0.245g(收率=62%)的澄清的黃色微晶固體產物。
元素分析[實測的(計算的)]:C:46.88%(47.32%);H:3.01%(3.30%);N:5.76%(6.13%);Zr:29.44%(19.96%);Cl:24.01%(23.27%)。
實施例15
ZrCl2(L5)[樣品MT-81]的合成
向100ml長頸燒瓶中引入如在實施例5中描述獲得的具有式(L5)的配體(0.38g;2.08mmol)在四氫呋喃(10ml)中的溶液:使整體冷卻到-70℃並且隨後逐滴地添加正丁基鋰(0.87ml,2.17mmol)在己烷中的溶液,以獲得黃-橙色懸浮液。將獲得的懸浮液加熱到室溫,並且留在此溫度下在攪拌下持續3小時。隨後,逐滴地添加四氯化鋯四氫呋喃(1:2)[ZrCl4(THF)2](0.391g;1.04mmol;L5/Zr摩爾比=2)在四氫呋喃(30ml)中的溶液:在添加第一個10ml之後,獲得橙色溶液,而在添加結束時,獲得黃色溶液,將所述黃色溶液留在室溫下在攪拌下持續一夜。隨後,將溶劑通過在減壓下在室溫下的蒸餾除去,以獲得黃色殘餘物,所述殘餘物用二氯甲烷(15ml)處理。將獲得的懸浮液過濾,並且將濾液濃縮到一半體積,用己烷(20ml)處理,並且保持在-30℃下持續一夜。隨後,將獲得的殘餘物通過過濾回收,用庚烷(2×1ml)洗滌並且在真空下在室溫下乾燥,以獲得對應於ZrCl2(L5)2絡合物的0.27g(收率=35%)的棕色微晶固體產物。
元素分析[實測的(計算的)]:C.53.79%(53.54%);H:3.89%(4.19%);N:10.99%(10.60%);Zr:18.01%(17.26%);Cl:12.98%(13.41%)。
實施例16(GL957)
向50ml試管中,在冷卻時(-20℃),冷凝等於約1.4g的2ml的1,3-丁二烯。隨後,添加4.65ml的甲苯並且使如此獲得的溶液的溫度達到20℃。隨後,添加在甲苯溶液中的甲基鋁氧烷(MAO)(15.75ml;2.5×10-2mol,等於約1.45g),隨後是如在實施例8中描述獲得的ZrCl3(L1)絡合物[樣品BM2-199](以5mg/ml的濃度的4.6ml的甲苯溶液;5×10-5mol,等於約23mg)。整體被保持在20℃下磁力攪拌持續6小時。隨後,聚合通過添加包含若干滴鹽酸的2ml甲醇被猝滅。隨後,獲得的聚合物通過添加包含4%的1076抗氧化劑(Ciba)的40ml甲醇溶液被凝結,以獲得具有96%的1,4-反式單元含量的0.97g的聚丁二烯:工藝和獲得的聚丁二烯的另外的特徵在表1中被示出。
圖6(a)示出獲得的聚丁二烯的FT-IR光譜。
實施例17(GL959)
向50ml試管中,在零下溫度(-20℃),冷凝等於約1.4g的2ml的1,3-丁二烯。隨後,添加4.45ml的甲苯並且使如此獲得的溶液的溫度達到20℃。隨後,添加在甲苯溶液中的甲基鋁氧烷(MAO)(15.75ml;2.5×10-2mol,等於約1.45g),隨後是如在實施例9中描述獲得的ZrCl3(L2)絡合物[樣品BM2-207](以5mg/ml的濃度的4.8ml的甲苯溶液;5×10-5mol,等於約24mg)。整體被保持在20℃下磁力攪拌持續7小時。隨後,聚合通過添加包含若干滴鹽酸的2ml甲醇被猝滅。隨後,獲得的聚合物通過添加包含4%的1076抗氧化劑(Ciba)的40ml甲醇溶液被凝結,以獲得具有等於95%的1,4-反式單元含量的0.63g的聚丁二烯:工藝和獲得的聚丁二烯的另外的特徵在表1中被示出。
圖6(b)示出獲得的聚丁二烯的FT-IR光譜。
圖7示出獲得的聚丁二烯的13C-NMR光譜。
圖12示出獲得的聚丁二烯的GPC曲線圖。
實施例18(MM20)
向第一個50ml試管中,在冷卻時(-20℃),冷凝等於約1.4g的2ml的1,3-丁二烯。隨後,添加10.1ml的甲苯並且使如此獲得的溶液的溫度達到20℃。隨後,添加在甲苯溶液中的幹甲基鋁氧烷(MAO幹)(10ml;3×10-2mol,等於約1.74g)。向第二個10ml試管中引入如在實施例9中描述獲得的ZrCl3(L2)絡合物[樣品BM2-207](以5mg/ml的濃度的2.9ml的甲苯溶液;3×10-5mol,等於約14.4mg)和三乙基鋁(以0.052mg/ml的濃度的2ml的甲苯溶液;9×104mol,等於約104mg):將整體保持在室溫下在攪拌下持續10分鐘,並且將獲得的溶液完全添加到所述第一個試管。整體被保持在20℃下磁力攪拌持續2小時。隨後,聚合通過添加包含若干滴鹽酸的2ml甲醇被猝滅。隨後,獲得的聚合物通過添加包含4%的1076抗氧化劑(Ciba)的40ml甲醇溶液被凝結,以獲得具有等於99%的1,4-反式單元含量的1.24g的聚丁二烯:工藝和獲得的聚丁二烯的另外的特徵在表1中被示出。
圖6(c)示出獲得的聚丁二烯的FT-IR光譜。
圖8示出獲得的聚丁二烯的1H-NMR光譜。
圖9示出獲得的聚丁二烯的13C-NMR光譜。
圖13示出獲得的聚丁二烯的GPC曲線圖。
圖18示出獲得的聚丁二烯的DSC曲線圖。
實施例19(G1125)
向第一個25ml試管中,在冷卻時(-20℃),冷凝等於約1.4g的2ml的1,3-丁二烯。隨後,添加9.3ml的甲苯並且使如此獲得的溶液的溫度達到20℃。隨後,添加在甲苯溶液中的幹甲基鋁氧烷(MAO幹)(10ml;3×10-2mol,等於約1.74g)。向第二個10ml試管中引入如在實施例10中描述獲得的ZrCl3(L3)絡合物[樣品MT-2](以5mg/ml的濃度的2.7ml的甲苯溶液;3×10-5mol,等於約13.4mg)和氫化二異丁基鋁(DIBAH)(以0.040mg/ml的濃度的3ml的甲苯溶液;8.4×10-4mol,等於約120mg):將整體保持在室溫下在攪拌下持續10分鐘,並且將獲得的溶液完全添加到所述第一個試管。整體被保持在20℃下磁力攪拌持續4小時。隨後,聚合通過添加包含若干滴鹽酸的2ml甲醇被猝滅。隨後,獲得的聚合物通過添加包含4%的1076抗氧化劑(Ciba)的40ml甲醇溶液被凝結,以獲得具有等於99%的1,4-反式單元含量的1.24g的聚丁二烯:工藝和獲得的聚丁二烯的另外的特徵在表1中被示出。
圖6(d)示出獲得的聚丁二烯的FT-IR光譜。
實施例20(G1112)
向25ml試管中,在冷卻時(-20℃),冷凝等於約1.4g的2ml的1,3-丁二烯。隨後,添加5.15ml的甲苯並且使如此獲得的溶液的溫度達到20℃。隨後,添加在甲苯溶液中的甲基鋁氧烷(MAO)(15.75ml;2.5×10-2mol,等於約1.45g),隨後是如在實施例12中描述獲得的ZrCl3(L6)絡合物[樣品MT-30](以5mg/ml的濃度的4.1ml的甲苯溶液;3×10-5mol,等於約20.5mg)。整體被保持在20℃下磁力攪拌持續7小時。隨後,聚合通過添加包含若干滴鹽酸的2ml甲醇被猝滅。隨後,獲得的聚合物通過添加包含4%的1076抗氧化劑(Ciba)的40ml甲醇溶液被凝結,以獲得具有等於95%的1,4-反式單元含量的0.68g的聚丁二烯:工藝和獲得的聚丁二烯的另外的特徵在表1中被示出。
圖10示出獲得的聚丁二烯的1H-NMR光譜。
圖11示出獲得的聚丁二烯的13C-NMR光譜。
實施例21(MM21)
向第一個25ml試管中,在冷卻時(-20℃),冷凝等於約1.4g的2ml的1,3-丁二烯。隨後,添加9.54ml的甲苯並且使如此獲得的溶液的溫度達到20℃。隨後,添加在甲苯溶液中的幹甲基鋁氧烷(MAO幹)(10ml;3×10-2mol,等於約1.74g)。向第二個10ml試管中引入如在實施例12中描述獲得的ZrCl3(L6)絡合物[樣品MT-30](以5mg/ml的濃度的2.7ml的甲苯溶液;3×10-5mol,等於約13.44mg)和三異丁基鋁(TIBA)(以0.056mg/ml的濃度的3ml的甲苯溶液;8.4×10-4mol,等於約167mg):將整體保持在室溫下在攪拌下持續10分鐘,並且將獲得的溶液完全添加到所述第一個試管。整體被保持在20℃下磁力攪拌持續4小時。隨後,聚合通過添加包含若干滴鹽酸的2ml甲醇被猝滅。隨後,獲得的聚合物通過添加包含4%的1076抗氧化劑(Ciba)的40ml甲醇溶液被凝結,以獲得具有等於99%的1,4-反式單元含量的1.15g的聚丁二烯:工藝和獲得的聚丁二烯的另外的特徵在表1中被示出。
圖6(e)示出獲得的聚丁二烯的FT-IR光譜。
圖14示出獲得的聚丁二烯的GPC曲線圖。
實施例22(G1120)
向25ml試管中,在冷卻時(-20℃),冷凝等於約1.4g的2ml的1,3-丁二烯。隨後,添加4.65ml的甲苯並且使如此獲得的溶液的溫度達到20℃。隨後,添加在甲苯溶液中的甲基鋁氧烷(MAO)(15.75ml;2.5×10-2mol,等於約1.45g),隨後是如在實施例14中描述獲得的ZrCl3(L4)絡合物[樣品MT-56](以5mg/ml的濃度的4.6ml的甲苯溶液;5×10-5mol,等於約23mg)。整體被保持在20℃下磁力攪拌持續6小時。隨後,聚合通過添加包含若干滴鹽酸的2ml甲醇被猝滅。隨後,獲得的聚合物通過添加包含4%的1076抗氧化劑(Ciba)的40ml甲醇溶液被凝結,以獲得具有等於95%的1,4-反式單元含量的0.55g的聚丁二烯:工藝和獲得的聚丁二烯的另外的特徵在表1中被示出。
圖15示出獲得的聚丁二烯的GPC曲線圖。
實施例23(G1121)
向25ml試管中,在冷卻時(-20℃),冷凝等於約1.4g的2ml的1,3-丁二烯。隨後,添加2.15ml的甲苯並且使如此獲得的溶液的溫度達到20℃。隨後,添加在甲苯溶液中的甲基鋁氧烷(MAO)(15.75ml;2.5×10-2mol,等於約1.45g),隨後是如在實施例15中描述獲得的ZrCl2(L5)2絡合物[樣品MT-81](以5mg/ml的濃度的5.3ml的甲苯溶液;5×10-5mol,等於約26.4mg)。整體被保持在20℃下磁力攪拌持續5小時。隨後,聚合通過添加包含若干滴鹽酸的2ml甲醇被猝滅。隨後,獲得的聚合物通過添加包含4%的1076抗氧化劑(Ciba)的40ml甲醇溶液被凝結,以獲得具有等於94%的1,4-反式單元含量的1.36g的聚丁二烯:工藝和獲得的聚丁二烯的另外的特徵在表1中被示出。
圖16示出獲得的聚丁二烯的GPC曲線圖。
圖17示出獲得的聚丁二烯的DSC曲線圖。
表1
使用包含鋯的絡合物的催化體系使1,3-丁二烯聚合
(a):熔點;
(b):結晶溫度。