用於高溫聚合的齊格勒‑納塔催化劑的製作方法
2023-11-11 22:08:52 4
本發明涉及用於聚烯烴的高溫溶液聚合的鎂-鈦催化劑。
背景技術:
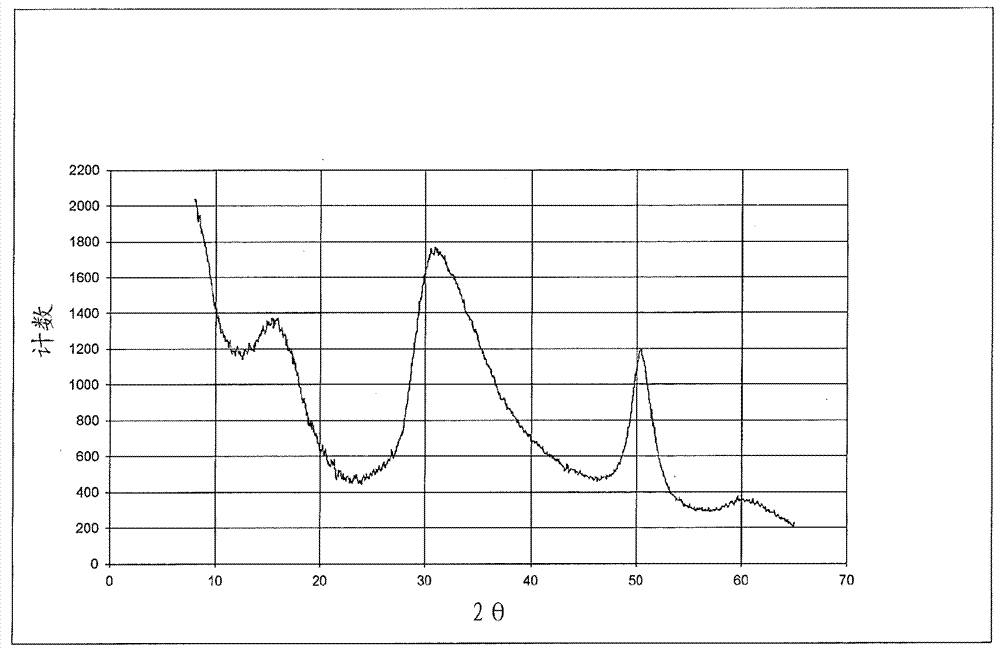
:用於烯烴聚合的鎂-鈦催化劑在商業上廣泛使用。通常而言,這些催化劑包含鎂滷化物組分(通常是二氯化鎂)和沉積於二氯化鎂上的鈦組分。所得鎂-鈦複合物常被稱為「主催化劑」,這是由於其需要助催化劑或活化劑來產生高度反應性的聚合催化劑體系。可以首先合成主催化劑,然後在之後添加至聚合反應器。或者,主催化劑可以通過「管線內(in-line)混合技術」(鄰近聚合反應器)來製備,並直接添加至反應器。許多原始的齊格勒-納塔催化劑活性不足以使催化劑殘留物能夠留在聚合物中而不引起品質問題(例如聚合物顏色、以及在不合需要的短時期內使聚合物降解/氧化的傾向)。相應地,存在對「高活性駐留式(leave-in)」催化劑的需求,其特徵在於,其具有較少的可留在成品聚合物中的會引起問題的催化劑殘留物。存在對用於聚烯烴的高溫溶液聚合的高度活性鎂-鈦催化劑的需求,其可以在較低的殘留鈦和滷素雜質的情況下提供增加的共聚單體引入和較高分子量的聚合物材料。技術實現要素:本發明的一些實施方案提供用於乙烯和α-烯烴的聚合的主催化劑,所述主催化劑包含至少0.2%的可通過epr檢測的具有1.950的g值的物質。本發明的一些實施方案提供用於乙烯和α-烯烴的聚合的在δ型mgcl2載體上的主催化劑,所述主催化劑包含式ticl3*[[r4]a[r5o]balx3-c]d的ti3+配合物,其中,a為0至1;b為0至1;c=a+b;d為約0.33至約1.0;每個r4和r5獨立地選自c1-8烷基;每個x獨立地選自滷素;且進一步地其中,存在的總ti中的至少60%呈ti3+氧化態。本發明的其他實施方案提供用來製備包含ti3+配合物的烯烴聚合主催化劑的方法,所述方法包括:a)通過組合以下物質來形成δ型mgcl2物質:i)選自c5-12烷烴的溶劑中的r2mg,和ii)反應性有機氯化物或hcl,其中,每個r獨立地選自c2-8烷基,並且其中所添加的cl和mg的摩爾比為約2.0至約3.0;b)向步驟a中製備的所述δ型mgcl2物質,以任意順序或同時添加r1xalx3-x和四價鈦化合物,其中,al/ti摩爾比為約3至約10;或者c)向步驟a中製備的所述δ型mgcl2物質,先添加r1xalx3-x,其次添加四價鈦化合物,接著添加r4yalor53-y,其中,當測量僅由r1xalx3-x供應的al時的al/ti摩爾比為約0.7至約2,並且當測量由r4yalor53-y供應的al時的al/ti摩爾比為約1至約2;且進一步地其中,mg/ti摩爾比為約5至約10;x為1或2;y為1或2;每個r1獨立地選自c1-8烷基;四價鈦化合物選自tir2x3、ti(or3)x3、tix4、和它們的混合物;每個x獨立地選自滷素;每個r2獨立地選自c1-8烷基和苄基,並且每個r3、r4和r5獨立地選自c1-8烷基。本發明的其他實施方案提供通過以下方法製備的包含ti3+配合物的主催化劑產物,所述方法包括:a)通過組合以下物質來形成δ型mgcl2物質:i)選自c5-12烷烴的溶劑中的r2mg,和ii)反應性有機氯化物或hcl,其中,每個r獨立地選自c2-8烷基,並且其中所添加的cl和mg的摩爾比為約2.0至約3.0;b)向步驟a中製備的所述δ型mgcl2物質,以任意順序或同時添加r1xalx3-x和四價鈦化合物,其中,al/ti摩爾比為約3至約10;或者c)向步驟a中製備的所述δ型mgcl2物質,先添加r1xalx3-x,其次添加四價鈦化合物,接著添加r4yalor53-y,其中,當測量僅由r1xalx3-x供應的al時的al/ti摩爾比為約0.7至約2,並且當測量由r4yalor53-y供應的al時的al/ti摩爾比為約1至約2;並且進一步地其中,mg/ti摩爾比為約5至約10;x為1或2;y為1或2;每個r1獨立地選自c1-8烷基;四價鈦化合物選自tir2x3、ti(or3)x3、tix4、和它們的混合物;每個x獨立地選自滷素;每個r2獨立地選自c1-8烷基和苄基,並且每個r3、r4和r5獨立地選自c1-8烷基。本發明的其他實施方案提供溶液烯烴聚合方法,其包括:i)向任選具有一個或多個附加反應器的連續攪拌釜式反應器(cstr),添加選自c5-12烷烴的溶劑和用於聚合的在δ型mgcl2載體上的主催化劑,所述主催化劑包含式ticl3*[[r4]a[r5o]balx3-c]d的ti3+配合物,其中,a為0至1;b為0至1;c=a+b;d為0.33至1.0;每個r4和r5獨立地選自c1-8烷基;每個x獨立地選自滷素;其中,存在的總ti中的至少60%呈ti3+氧化態;ii)向反應器添加乙烯、氫氣和任選一種或多種選自c3-8共聚單體的共聚單體;和iii)向反應器以相對於主催化劑的量為約1至約10的摩爾比添加烷基鋁活化劑。本發明的其他實施方案提供通過以下聚合方法製備的烯烴聚合產物,所述方法包括:i)向任選具有一個或多個附加反應器的連續攪拌釜式反應器(cstr),添加選自c5-12烷烴的溶劑和用於聚合的在δ型mgcl2載體上的主催化劑,所述主催化劑包含式ticl3*[[r4]a[r5o]balx3-c]d的ti3+配合物,其中,a為0至1;b為0至1;c=a+b;d為0.33至1.0;每個r4和r5獨立地選自c1-8烷基;每個x獨立地選自滷素;其中,存在的總ti中的至少60%呈ti3+氧化態;ii)向反應器添加乙烯、氫氣和任選一種或多種選自c3-8共聚單體的共聚單體;和iii)向反應器以相對於主催化劑的量為約1至約10的摩爾比添加烷基鋁活化劑。本發明的其他實施方案提供選自膜、纖維、模塑或熱成型的製品、以及管道塗層的塑料製品,其包含通過下述聚合方法製備的烯烴聚合產物,所述方法包括:i)向任選具有一個或多個附加反應器的連續攪拌釜式反應器(cstr),添加選自c5-12烷烴的溶劑和用於聚合的在δ型mgcl2載體上的主催化劑,所述主催化劑包含式ticl3*[[r4]a[r5o]balx3-c]d的ti3+配合物,其中,a為0至1;b為0至1;c=a+b;d為0.33至1.0;每個r4和r5獨立地選自c1-8烷基;每個x獨立地選自滷素;其中,存在的總ti中的至少60%呈ti3+氧化態;ii)向反應器添加乙烯、氫氣和任選一種或多種選自c3-8共聚單體的共聚單體;和iii)向反應器以相對於主催化劑的量為約1至約10的摩爾比添加烷基鋁活化劑。附圖簡述圖1示出來自α型mgcl2的典型xrd譜。圖2示出使用在本文中公開並要求保護的方法形成的δ型mgcl2的xrd圖譜。圖3示出產物5的gpc-ft-ir。圖4示出催化劑1的epr譜和模擬。圖5示出催化劑2b的epr譜和模擬。圖6示出對比實施例a的epr譜和模擬。具體實施方式除在操作實施例中或在另有指明之處外,所有在說明書和權利要求書中使用的指代成分的量、反應條件等的數字或表達均要被理解為在所有情況下被術語「約」修飾。相應地,除非有相反指明,在以下說明書和隨附的權利要求書中陳述的數值參數是可以根據本發明期望獲得的所需特性而變化的近似值。在最低限度上,並且不試圖限制將等同原則應用於權利要求的範圍,應當至少根據所報導的有效數字的數值並且通過應用普通捨入技術來解釋各數值參數。儘管陳述本發明的寬泛範圍的數值範圍和參數是近似值,但在具體實施例中陳述的數值則儘可能精確地報導。但任何數值都固有地包含一定的由在其各自的測試測量中存在的標準偏差所必然導致的誤差。同時,應當理解的是,在本文中引述的任何數值範圍旨在包括在其中包含的所有子範圍。例如,範圍「1至10」旨在包括之間的全部子範圍並包括所引述的最小值1和所引述的最大值10;也即是說,具有等於或大於1的最小值和等於或小於10的最大值。由於所公開的數值範圍是連續的,其包括在最小值和最大值之間的每個值。除非另有明確指明,在本申請中指定的多個數值範圍是近似值。在本文中表達的所有組成範圍在實踐中限制在總計100%且不超過100%(體積%或重量%)。在組合物中可以存在多種組分的情況下,各組分的最大量的總和可以超過100%,要理解且本領域技術人員容易理解的是,實際使用的組分的量將符合100%的最大值。必須注意的是,如在本文中和在隨附的權利要求中所使用的單數形式「一個」、「一種」和「所述」包括複數指代,除非上下文明確地另有規定。除非在本文中定義,在本文中使用的所有技術、科學術語具有與本發明所屬領域的普通技術人員所通常理解的相同含義。術語「烷基」、「烷基基團」和「烷基原子團」可以互換使用,並且是指鍵合至一個或多個其他部分的飽和的單價直鏈或支鏈和環狀的烴基基團或原子團。例如,烷基可以鍵合至氧原子從而形成烷氧基,或者作為在金屬上的配體的一部分或者作為配體而鍵合至金屬。術語「烷基」由例如以下基團例示:甲基、乙基、正丙基、異丙基、正丁基、叔丁基、正戊基、金剛烷基、環丙基、環丁基、環戊基、環己基、環辛基等。術語「烷烴」是指具有通式cnh(2n+2)的非芳族飽和烴分子,其中n為整數。烷烴例如可被用作溶劑或氣體進料。當在術語之前為cx-y(其中x和y為整數)時,所述基團限於在基團內x至y個碳原子,不包括被稱為取代基團的任何取代基。例如,c1-5烷基將包括(但不限於)甲基、異丙基、正丁基、叔丁基、環丙基和環戊基,而c1-5烷烴將包括(但不限於)甲烷、乙烷、戊烷、環戊烷等。術語「滷素原子團」或「滷素」或「滷代」可以互換使用,並且是指氟基團(fluoridegroup)、氯基團(chloridegroup)、溴基團(bromidegroup)或碘基團(iodidegroup)。主催化劑在一個實施方案中,在本文中描述的本發明是用於乙烯和α-烯烴的聚合的在δ型的mgcl2載體上的主催化劑,所述主催化劑包含式ticl3*[[r4]a[r5o]balx3-c]d的ti3+配合物,其中,a為0至1;b為0至1;c=a+b;d為0.33至1.0;每個r4和r5獨立地選自c1-8烷基;每個x獨立地選自滷素;並且其中,存在的總ti中的至少60%呈ti3+氧化態。儘管x可以為任意滷素,但在一些實施方案中,x為br或cl。在其他實施方案中,x為cl。在一些實施方案中,c為0。在其他實施方案中,c為1。在一些實施方案中,a為0並且b為1。在一些實施方案中,a為1且b為0。在一些實施方案中,a為1且b為1。在一些實施方案中,a為0且b為0。在一些實施方案中,每個r5為c1-4烷基。在其他實施方案中,每個r5為乙基。在一些實施方案中,每個r4為c1-4烷基。在其他實施方案中,每個r4為乙基。鎂/鈦摩爾比將要被鎂-鈦聚合催化劑領域的技術人員認識到的是,催化劑活性可受到鎂/鈦摩爾比的影響。對於本發明的催化劑,優選的摩爾mg/ti比為5/1至10/1,即催化劑中每摩爾的ti優選存在5至10摩爾的mg。在一些實施方案中,mg/ti摩爾比為約5至約8。在其他實施方案中,mg/ti比為約6至約8。所需mg/ti摩爾比可以通過根據本文中所描述的方法來製備主催化劑而獲得。主催化劑式和在其中包含的元素比率可以使用標準元素分析技術來確定,所述標準元素分析技術包括但不限於經典的「溼法化學」、中子活化、電感耦合等離子質譜(icp-ms)和x射線衍射譜(xrd)。針對鈦價態分布,可以使用針對鈦價態分布的氧化還原滴定法(參見j.c.w.chien等人,j.polym.sci.parta:polymchem.1989,27,1499-1514)來分析催化劑樣品,或針對鈦含量分析,基於astm標準e878-01使用紫外(uv)法來分析催化劑樣品。在一些實施方案中,存在的總ti中的至少70%呈ti3+氧化態。在其他實施方案中,存在的總ti中的至少80%呈ti3+氧化態。固體齊格勒催化劑的表徵可以通過電子順磁共振譜(epr)來實現,呈氧化態+3的鈦原子的部分對其靈敏。g值歸屬基於出版物j.c.w.chien等人,j.polym.sci.parta:poly.chem.1982,20,2461-2476。在檢驗在本文中描述的齊格勒催化劑的epr譜和它們相應的模擬譜時,觀察到三組epr峰,並將其歸屬於物質a、b和c。具有1.910、1.898、1.955的g值的物質a據信為其中來自mgcl2的兩個氯離子配位至ticl3的物質(該配合物據信類似於在chien(1982)參考文獻中歸屬於物質a的配合物)。物質b由於峰非常寬而難以界定。當物質c以1.950的g值存在時,其據信為其中來自mgcl2的單個cl配位至al的物質(該配合物據信類似於在chien(1982)參考文獻中歸屬於物質f的配合物);而當物質c存在並改為具有1.969的g值時,其據信為其中來自mgcl2的單個cl配位至ti的物質(該配合物據信類似於在chien(1982)參考文獻中歸屬於物質e的配合物)。在一個實施方案中,本文中描述的主催化劑的固體組分包含至少0.2%、或例如約0.2%至約1%的具有1.950的g值的物質c,就在圖4和5中所說明的意義而言。據信,該物質c在ti處具有四面體構型。在其他實施方案中,主催化劑具有約0.2%至約0.5%的具有1.950的g值的物質c,或者具有約0.5%至約1%的具有1.950的g值的物質c。相比之下,對比實施例a的固體組分使用其他的已知製備方法和在美國專利第7,666,810b2號中描述的方法來獲得。對比實施例a的epr分析和模擬顯示出存在具有1.969的g值的物質c(示於圖6中),其可以歸屬為在ti處的三角雙錐構型。不希望被束縛於任何特定的解釋理論,已發現,當物質c以1.969的g值存在時,主催化劑與物質c以1.950的g值存在時的主催化劑相比,展現出在烯烴聚合活性方面和在高溫聚合方法中生產的聚合物的分子量方面的更不利的特性。在一個實施方案中,主催化劑為式ticl3*[oetalcl2]d的ti3+配合物,並且mg/ti摩爾比為約5至約8。在另一個實施方案中,主催化劑為式ticl3*[clalcl2]d的ti3+配合物,並且mg/ti比為約5至約8。在一些實施方案中,可以存在ticl3*[clalcl2]d或ticl3*[oetalcl2]d的部分烷基化形式。在本文中描述的本發明的另一個實施方案提供用來製備包含ti3+配合物的烯烴聚合主催化劑的方法,所述方法包括:a)通過組合以下物質來形成δ型mgcl2物質:i)選自c5-12烷烴的溶劑中的r2mg,和ii)反應性有機氯化物或hcl;其中,每個r獨立地選自c2-8烷基;並且其中所添加的cl與mg的摩爾比為約2.0至約3.0;然後,可替代地b)向步驟a)中製備的所述δ型mgcl2物質,以任意順序或同時添加式r1xalx3-x的烷基鋁滷化物和四價鈦化合物,從而提供約3至約10的al/ti摩爾比;或者c)向步驟a)中製備的所述δ型mgcl2物質,首先添加式r1xalx3-x的烷基鋁滷化物,並其次添加四價鈦化合物,然後在最終添加步驟中添加式r4yalor53-y的烷基鋁烷氧化物,其中,當測量由r1xalx3-x供應的al時的al/ti摩爾比為約0.7至約2,並且當測量由r4yalor53-y供應的al時的al/ti摩爾比為約1至約2;並且進一步地其中,mg/ti摩爾比為約5至約10,x為1或2,y為1或2,每個r1獨立地選自c1-8烷基,四價鈦化合物選自tir2x3、ti(or3)x3、tix4、和它們的混合物,每個x獨立地選自滷素,每個r2獨立地選自c1-8烷基和苄基,並且每個r3、r4和r5獨立地選自c1-8烷基。在本文中描述的本發明的另一個實施方案提供通過以下方法製備的主催化劑產物,所述方法包括:a)通過組合以下物質來形成δ型mgcl2物質:i)選自c5-12烷烴的溶劑中的r2mg,和ii)反應性有機氯化物(rcl)或hcl;其中,每個r獨立地選自c2-8烷基;並且其中所添加的cl與mg的摩爾比為約2.0至約3.0;然後,可替代地b)向步驟a)中製備的所述δ型mgcl2物質,以任意順序或同時添加式r1xalx3-x的烷基鋁滷化物和四價鈦化合物,其中,al/ti摩爾比為約3至約10;或者c)向步驟a)中製備的所述δ型mgcl2物質,首先添加式r1xalx3-x的烷基鋁滷化物,並其次添加四價鈦化合物,然後在最終添加步驟中添加式r4yalor53-y的烷基鋁烷氧化物,其中,當測量由r1xalx3-x供應的al時的al/ti摩爾比為約0.7至約2,並且當測量由r4yalor53-y供應的al時的al/ti摩爾比為約1至約2;並且進一步地其中,mg/ti摩爾比為約5至約10,x為1或2,y為1或2,每個r1獨立地選自c1-8烷基,四價鈦化合物選自tir2x3、ti(or3)x3、tix4、和它們的混合物,每個x獨立地選自滷素,每個r2獨立地選自c1-8烷基和苄基,並且每個r3、r4和r5獨立地選自c1-8烷基。二有機鎂二有機鎂化合物是公知且商業上可獲得的。二有機鎂化合物通常可以由式mgr2表示,其中,每個r選自c2-8烴基。在一個實施方案中,每個r獨立地選自直鏈c2-8烷基,包括但不限於乙基、丁基、己基和辛基。在另一個實施方案中,每個r獨立地選自c2-4烷基。在另一個實施方案中,每個r獨立地選自乙基和丁基。在一個實施方案中,mgr2選自丁基乙基鎂(bem)、二丁基鎂、和丁基辛基鎂(bom)。在另一個實施方案中,mgr2是丁基乙基鎂(bem)。二有機鎂溶液是由albemarle銷售的商業上可獲得的材料。其他二有機鎂化合物包括丁基乙基鎂或二丁基鎂的烴溶液(其可任選用有機鋁化合物處理以改進溶解性和/或降低溶液粘度)。在一個實施方案中,mgr2提供於選自c5-12烷烴的溶劑中。在一個實施方案中,溶劑選自己烷、環己烷、癸烷、庚烷、異己烷、和十二烷、以及它們的混合物。在一個實施方案中,溶劑為異己烷。在一個實施方案中,溶劑為癸烷。在一個實施方案中,溶劑為庚烷。氯的量和氯源是在「鎂-鈦」聚合催化劑中使用二氯化鎂是公知的。mgcl2通常被當做鈦物質的載體。二有機鎂化合物與2摩爾當量的氯反應以製備二氯化鎂是用來製備催化劑載體的公知方法。本發明的實施方案使用二氯化鎂載體,其通過二有機鎂化合物(上文所述)與2至3摩爾當量的氯的反應來製備。在一個實施方案中,載體中的氯/鎂比率為每摩爾鎂約2.15至約3.0(基於起始二有機鎂化合物中的鎂的量)、或者約2.15至約2.5。氯源基本上自發地與二有機鎂反應,並且是反應性的有機氯化物或hcl。在一個實施方案中,反應性的有機氯化物為c4-10叔烷基氯化物。在一個實施方案中,反應性的有機氯化物為叔丁基氯。在一個實施方案中,氯源為hcl。反應溫度可為約20℃至約160℃、或者約40℃至約100℃、或者約50℃至90℃、或者約40℃至約90℃。按本文中公開來製備的mgcl2物質呈δ型,如通過測量x射線衍射測量的峰的半高度而確定。本領域技術人員已知δ型是α和β型mgcl2的高度無序的混合物。xrd譜在確定mgcl2載體的結構方面特別有用,所述結構通過通常以旋轉變換無序(rototranslationaldisorder)為特徵的結構的x射線譜來表徵(參見例如g.natta等人,j.polym.sci.1961,51,399-410)。圖1示出典型的來自α型的mgcl2的xrd譜。圖2示出使用在本文中公開並要求保護的方法形成的δ型mgcl2的xrd圖譜。在本文中描述的本發明的一些實施方案中,用於製備mgcl2物質的方法的優點使得主催化劑形成的下一步驟能夠繼續進行而不需要介入洗滌步驟(如果需要該步驟)。殘留的二有機鎂起始材料的有害影響通過以下方式而最小化:使起始材料反應從而達到所公開的cl與mg的摩爾比,或者用額外的氯源、例如異丁基alcl2來處理mgcl2。鈦iv源然後,通過在上文描述的氯化鎂載體上沉積鈦化合物來製備本文中描述的主催化劑。起始鈦(iv)化合物可以選自式tir2x3、ti(or3)x3、tix4的化合物,和它們的混合物,其中,每個r2選自c1-8烷基和苄基,並且r3選自c1-8烷基,並且每個x獨立地為滷素。在一些實施方案中,滷素選自氯和溴。在其他實施方案中,滷素是氯。在一些實施方案中,r3選自c1-4烷基。在其他實施方案中,r3選自乙基、異丙基和叔丁基。在一些實施方案中,r2選自c1-4烷基。在其他實施方案中,r2選自乙基和異丁基。在一些實施方案中,r2為苄基。在一些實施方案中,四價鈦化合物為ti(och2ch3)cl3、或ti(ch2ch3)cl3。在一些實施方案中,四價鈦化合物選自ticl2br2和ticl4。在一些實施方案中,四價鈦化合物為ticl4。將要被本領域技術人員理解的是,tir2x3、ti(or3)x3、tix4物質可以購買,或者替代地可以通過用公知的商業上可獲得且廉價的烷基鈦和烷氧基鈦化合物反應來製備,所述烷基鈦和烷氧基鈦化合物例如為ti(r2)2x2、ti(r2)3x1、ti(or3)2x2、或ti(or3)3x1,其中,各個x、r2和r3如上文中所述。鋁物質在本文中描述的方法中所使用的鋁化合物在商業上購買於公司例如albemarle、sigma-aldrich、或fisherchemical。r1xalx3-x用於滷化二烷基鎂化合物和格式試劑,並且以在上文中指定的摩爾比量進行添加從而使溶液中的過量滷素最小化並且使ti物質的過度還原最小化。在一些實施方案中,x為1。在其他實施方案中,x為2。在一些實施方案中,每個r1獨立地選自甲基、乙基、丙基、異丙基、丁基和異丁基。在其他實施方案中,每個r1獨立地為乙基和異丁基。儘管x可以為任意滷素,在一些實施方案中,x為cl或br。在其他實施方案中,x為cl。在本文中描述的製造主催化劑的方法的一個實施方案中,r1xalx3-x選自異丁基二氯化鋁(ibadc)和乙基二氯化鋁(eadc)。r4yalor53-y用於將鈦物質還原成所需氧化態和/或可以與過量的滷化物反應。除此之外,該化合物可以充當用於在本文下文中公開的聚合反應的活化劑。除了r4yalor53-y物質之外,還可以使用上文描述的r1xalx3-x作為還原劑。其他還原劑包括alr*3、alr*2x、至alr*1x2,其中,r*為c2-8烷基。儘管r*可以為更高級的烷基,但這樣的鋁物質在商業上不合需要。在本文中描述的製造主催化劑的方法的一些實施方案中,r1xalx3-x為三異丁基鋁。在一些實施方案中,y為2。在一些實施方案中,y為1。在一些實施方案中,每個r4和r5獨立地選自c1-4烷基。在其他實施方案中,每個r4和r5為乙基。在本文中描述的製造主催化劑的方法的一個實施方案中,r4yalor53-y為二乙基乙醇鋁(deal-e)。通過後續向mgcl2物質添加鋁和鈦物質來進行的主催化劑的製備可以通過替代的路徑實現。在一個實施方案中,使用以任意順序添加至鈦化合物或者與鈦化合物一起添加的r1xalx3-x化合物實現鈦物質從ti4+至ti3+的還原。在該路徑的一些實施方案中,al/ti摩爾比為約4至7。在該路徑的其他實施方案中,al/ti比為約5。在另一個替代的路徑中,在添加較少量的r1xalx3-x化合物(與在先前討論的路徑中使用的r1xalx3-x化合物的量相比)後,添加鈦物質。還原為ti3+物質通過添加r4yalor53-y化合物來完成。在該路徑的一些實施方案中,當測量由r1xalx3-x供應的al時的al/ti摩爾比為約1至約1.8。在該路徑的其他實施方案中,當測量由r1xalx3-x供應的al時的al/ti摩爾比為約1。在該路徑的一些實施方案中,當測量由r4yalor53-y供應的al時的al/ti摩爾比為約0.7至約1.7、或者約1.5至1.7。在該路徑的其他實施方案中,當測量由r4yalor53-y供應的al時的al/ti摩爾比為約1.67。在所討論的任一路徑中,反應可以在約40℃至90℃、或者約40℃至約70℃、或者約45℃至約55℃的溫度下進行,或者在約50℃的溫度下進行。電子供體使用電子供體在基於鎂-鈦的烯烴聚合催化劑的領域中是公知的。本發明涵蓋對電子供體的任選使用。但是,當在溶液聚合條件下使用催化劑時,優選不使用電子供體。適合的電子供體對本領域技術人員而言是公知的,並且包括四氫呋喃(thf)、二甲基甲醯胺、乙酸乙酯、甲基異丁酮和各種鄰苯二甲酸酯/鹽。活化劑可以在本發明中使用任何的「活化劑」,其活化上文描述的用於烯烴聚合的鎂/鈦主催化劑。示例性的活化劑包括鋁氧烷和有機鋁助催化劑。鋁氧烷可以具有下式:(r6)2alo(r6alo)mal(r6)2其中,每個r6獨立地選自c1-20烴基,並且m為0至50,優選r6為c1-4烷基,並且m為5至30。甲基鋁氧烷(或「mao」)是優選的鋁氧烷,其中每個r6為甲基。鋁氧烷作為助催化劑,特別是用於茂金屬型催化劑的助催化劑是公知的。鋁氧烷也是易於獲得的商業製品。使用鋁氧烷助催化劑通常需要催化劑中的鋁與過渡金屬的摩爾比為25:1至1000:1。在本文中公開的方法中可用的示例比率為5:1至10:1。優選的有機鋁化合物包括三乙基鋁、三異丁基鋁和二乙基乙醇鋁。當使用這些有機鋁活化劑時,基於主催化劑中的ti的摩爾數而言,示例性的al/ti比為0.5/1至10/1。溶液聚合法優選以相對低的al/ti摩爾比(例如0.5/1至5/1,特別是1/1至3/1)來進行,而氣相聚合優選以相對高的al/ti摩爾比(例如20/1至150/1)來進行。用於乙烯的聚合和共聚的溶液法是本領域公知的。這些方法在惰性烴溶劑的存在下進行,所述惰性烴溶劑通常為c5-12烴,其可以未被取代或者被c1-4烷基取代,其例如為戊烷、甲基戊烷、己烷、庚烷、辛烷、環己烷、甲基環己烷和氫化石腦油。可商業上獲得的合適溶劑的一個實例是「isopare」(c8-12脂族溶劑,exxonchemicalco.)。常規淤漿法或溶液法中的聚合溫度為約80至約300℃(優選對於淤漿聚合而言為約80至約120℃,並且對於溶液聚合而言為約120至約250℃)。但是,如在實施例中例示的,用於本文中公開的溶液法的聚合溫度可以高於160℃。溫度上限將受到對本領域技術人員而言公知的考量的影響,例如期望使操作溫度最大化從而降低溶液粘度,同時仍保持良好的聚合物特性。提高的聚合溫度通常降低聚合物的分子量。在其他實施方案中,聚合溫度可以為約200至約300℃,或者約220至約250℃。反應工藝的一個實例是「中壓工藝」,其表示反應器中的壓力優選小於約6,000psi(約42,000千帕斯卡或kpa)。壓力可以為約10,000至約40,000kpa、或者約2,000至約3,000psi(約14,000-約22,000kpa)、或者725至約3,000psi(約5,000-約22,000kpa)。用於與乙烯共聚的合適單體包括c3-20單烯烴和二烯烴。示例共聚單體包括未被取代或者被最多兩個c1-6烷基取代的c3-12α-烯烴、未被取代或被最多兩個選自c1-4烷基的取代基取代的c8-12乙烯基芳族單體、未被取代或者被c1-4烷基取代的c4-12直鏈或環狀二烯烴。這樣的α-烯烴的說明性而非限制性的實例為以下中的一種或多種:丙烯、1-丁烯、1-戊烯、1-己烯、1-辛烯和1-癸烯、苯乙烯、α-甲基苯乙烯、和受限環(constrained-ring)環狀烯烴,例如環丁烯、環戊烯、二環戊二烯、降冰片烯、烷基取代的降冰片烯、烯基取代的降冰片烯等(例如5-亞甲基-2-降冰片烯和5-乙叉基-2-降冰片烯、雙環-(2,2,1)-庚-2,5-二烯)。乙烯與一種或多種可共聚單體的共聚物或三元共聚物還可以使用在本文中描述的方法來製備。在一個實施方案中,這樣的聚合物將含有約50至約75重量%的乙烯、優選約50至60重量%的乙烯、以及相應的50至40重量%的丙烯。一部分單體、通常為丙烯單體可以被共軛二烯烴替代。二烯烴可以多至聚合物的10重量%的量存在,儘管通常以約3至5重量%的量存在。所得聚合物可以具有包含40至75重量%的乙烯、50至15重量%的丙烯、和多至10重量%的二烯單體的組成以提供100重量%的聚合物。二烯的優選但非限制性的實例是二環戊二烯、1,4-己二烯、5-亞甲基-2-降冰片烯、5-乙叉基-2-降冰片烯、和5-乙烯基-2-降冰片烯,特別是5-乙叉基-2-降冰片烯和1,4-己二烯。在另一個實施方案中,所得聚合物可以包含不小於約80重量%、或不小於約90重量%的乙烯,和最多約20重量%、或小於10重量%的一種或多種可共聚單體。在一些實施方案中,共聚單體為c3-8α-烯烴,例如1-丁烯、1-己烯和1-辛烯。單體在被進料至反應器之前溶解/分散在溶劑中(或者對於氣態單體,可將單體進料至反應器使得其將溶解在反應混合物中)。在混合之前,可以對溶劑和單體進行純化從而除去潛在的催化劑毒性物,例如水、氧氣和其他極性雜質。原料純化遵照本領域中的標準實踐,例如使用分子篩、氧化鋁床和除氧催化劑以純化單體。優選還對溶劑本身(例如甲基戊烷、環己烷、己烷或甲苯)以類似方式進行處理。可以將原料在進料至反應器之前加熱或冷卻。在一些實施方案中,可以將催化劑組分預混合在用於反應的溶劑中,或者將其作為單獨的物流進料至反應器。在一些情況中,將其預混合可能是合乎需要的從而在進入反應之前為催化劑組分提供反應時間。本文中描述的本發明的一個實施方案提供溶液烯烴聚合方法,其包括:i)使用上文中描述的方法來製備主催化劑;ii)將主催化劑與選自c5-12烷烴的溶劑添加至呈串聯或並聯配置的一個或多個反應器,連同向反應器添加乙烯和任選的選自c3-8共聚單體的一種或多種共聚單體、氫氣;和iii)以相對於主催化劑的量為約1至約10的摩爾比將烷基鋁活化劑添加至反應器。聚合方法還可以使用選自r4yalor53-y、三烷基鋁化合物和mao的烷基鋁活化劑。在一些實施方案中,在聚合方法中使用的溶劑選自己烷、環己烷、癸烷、庚烷、異己烷和十二烷。在其他實施方案中,溶劑為異己烷。在其他實施方案中,溶劑為癸烷。在一些實施方案中,溶液法在單個連續攪拌釜式反應器(cstr)中並任選以一個或多個附加的反應器來進行。在其他實施方案中,溶液法在以串聯或並聯形式安裝的雙反應器式連續反應器中進行。本發明的方法還可以包括使用與至少一個cstr的排出物相連的管式反應器。(為清楚起見,如果串聯使用兩個cstr,則管式反應器接收來自第二cstr的排出物)。術語「管式反應器」意在表達其常規含義——即簡單的管。管式反應器可以具有至少10/1的長度/直徑(l/d)比。管式反應器不受攪動,並且以絕熱形式操作。因此,隨著聚合進行,剩餘的共聚單體越來越被消耗,並且溶液的溫度升高(這兩者改進了將剩餘的共聚單體與聚合物溶液分離的效率)。沿著管式反應器的長度的溫度增加可以大於3℃(即,來自管式反應器的排出物溫度比來自對管式反應器進料的cstr的排出物溫度大至少3℃)。管式反應器可以具有用於額外的乙烯和溶劑的進料口。進料為「經調溫的」——即額外的乙烯和/或溶劑的溫度被加熱至高於環境(或者至約100℃),但所述溫度低於管式反應器的排出物溫度。在一個實施方案中,乙烯被調溫至約80℃至約200℃、或者約100℃至約200℃。在一個實施方案中,與溶劑一起添加乙烯。溶劑的量(表述為基於乙烯的重量比)為約20/1至約0.1/1、或約10/1至約1/1。任選地,管式反應器還可以具有用於額外的催化劑、助催化劑、共聚單體和/或調聚劑(telomerizationagent)(例如氫氣)的進料口。但是,在一些實施方案中,不向管式反應器添加額外的催化劑。管式反應器的總體積可以為所述至少一個cstr的體積的至少10體積%,或者約30%至約200%(為清楚起見,如果cstr的體積為約1000升,則管式反應器的體積為至少約100升,或約300至約2000升)。添加至管式反應器的乙烯的總量可以為添加至一個或多個cstr的總乙烯的1至50重量%。例如,如果以約1000kg/hr的乙烯流量操作一個cstr,則至管式反應器的乙烯流將為約10至約500kg/hr。類似地,如果以約1000kg/hr的至第一cstr的乙烯流量,和約500kg/hr的至第二cstr的乙烯流量操作兩個cstr,則至管式反應器的乙烯流將為約15至約750kg/hr。在一些實施方案中,主催化劑是預配製的並且直接添加至反應器。在一些實施方案中,聚合溫度為至少約220℃、或至少約230℃、或至少約240℃。在一些實施方案中,與使用專利美國專利第5589555號中公開的主催化劑的聚合方法相比,使用本文中描述的主催化劑的聚合方法產生具有相同密度的聚合物,但其中所述方法使用少至少約10%的共聚單體進料。在一些實施方案中,與使用不包含至少0.2%的具有1.950的g值的epr活性物質的主催化劑的聚合方法相比,使用本文中描述的主催化劑的聚合方法產生具有相同密度的聚合物,但其中所述方法使用少至少約10%的共聚單體進料。在一些實施方案中,與使用不含四面體ti3+物質、或者基本不含四面體ti3+物質的用於聚合的主催化劑的聚合方法相比,使用本文中描述的主催化劑的聚合方法產生具有相同密度的聚合物,但其中所述方法使用少至少約10%的共聚單體進料。基本上不含四面體ti3+物質意指存在少於約0.005%、或少於0.01%、或少於0.05%的四面體ti3+物質,如通過如本文中描述的epr和epr模擬所確定的。在其他實施方案中,針對使用美國專利第5589555號中公開的主催化劑的製備的聚合物,使用本文中描述的主催化劑的聚合方法產生具有相同密度,但在任意聚合溫度下具有比前者聚合物所獲得的mw更高的mw的聚合物。在其他實施方案中,針對使用不包含至少0.2%的具有1.950的g值的epr活性物質的主催化劑製備的聚合物,使用本文中描述的主催化劑的聚合方法產生具有相同密度,但在任意聚合溫度下具有比前者聚合物所獲得的mw更高的mw的聚合物。在其他實施方案中,針對使用不含四面體ti3+物質、或者基本不含四面體ti3+物質的用於聚合的主催化劑製備的聚合物,使用本文中描述的主催化劑的聚合方法產生具有相同密度,但在任意聚合溫度下具有比前者聚合物所獲得的mw更高的mw的聚合物。基本上不含四面體ti3+物質意指存在少於約0.005%、或少於0.01%、或少於0.05%的ti3+物質,如通過如本文中描述的epr和epr模擬所確定的。在一些實施方案中,反應器保持時間(hold-uptime)為約30秒至約1小時。在其他實施方案中,反應器保持時間為約30秒至約30分鐘。在其他實施方案中,反應器保持時間為約30秒至約5分鐘。在其他實施方案中,反應器保持時間為約1分鐘至約5分鐘。本發明的另一個實施方案提供具有約0.910g/cc至約0.935g/cc的密度的聚乙烯聚合物或共聚物。本發明的另一個實施方案提供大於或等於約50的cdbi50辛烯。本發明的另一個實施方案提供具有約3至約8的mwd的聚合物。本發明的又一個實施方案在最終聚合物產物中提供基本上平坦(flat)的共聚單體分布。基本上平坦的共聚單體分布意指如在gpc曲線上描繪的作為分子量的函數的支鏈含量描點將給出偏離水平不大於約15°的線。在一些實施方案中,聚合物在所得聚合物中具有少於約10ppm的計算殘留鈦。在其他實施方案中,聚合物在所得聚合物中具有少於約8ppm的計算殘留鈦。在其他實施方案中,聚合物在所得聚合物中具有少於約3ppm的計算殘留鈦。在一些實施方案中,聚合物在所得聚合物中具有少於約120ppm的計算殘留滷素。在其他實施方案中,聚合物在所得聚合物中具有少於約100ppm的計算殘留滷素。在其他實施方案中,聚合物在所得聚合物中具有少於約60ppm的計算殘留滷素。本發明的另一個實施方案提供如在上文中描述的聚合物,其用於選自擠出、注射成型、熱成型、和旋轉成型的製造方法。本發明的另一個實施方案提供如在上文中描述的聚合物,其用於塑料製品,例如膜、纖維,模塑或熱成型的製品,例如鼓狀物和農業噴霧罐,以及管道塗層。將進一步通過參照以下實施例描述本發明。以下實施例僅是對本發明的例示,並且不意圖進行限制。除非另有指明,所有百分比以重量計。實施例化學品和試劑將購買的環己烷通過使其經過脫氧催化劑床(品牌名r311,來自basf)、氧化鋁床(品牌名selexsorbcos/cd)、和分子篩(3a/13x)床開進行乾燥和脫氧。正癸烷購買自sigmaaldrich,並將溶劑轉移至容納有經活化的13x分子篩的nalgene瓶中,並在使用之前儲存至少過夜。甲基戊烷購買自imperialoil,並且其包含100%的石油腦(石油)、經加氫處理的輕餾分(hydrotreatedlight)。通過使溶劑經過含有selexsorbcd和selexsorbcdx的床來將其乾燥。丁基乙基鎂(bem)(20wt.%在庚烷中的溶液)購買自albemarle。其容納於pyrosafe筒中,並且儲存在手套箱內。異丁基二氯化鋁(ibadc)(97wt.%)購買自albemarle。其容納於pyrosafe筒中,並且儲存在手套箱內。ibadc具有242℃的沸點和1.12g/ml的密度。二乙基乙醇鋁(deao)(25wt.%在庚烷中的溶液)購買自akzonobel。deao具有98℃的沸點和0.684g/ml的密度。乙基二氯化鋁(eadc)(20wt.%在庚烷中)購買自akzonobel。eadc具有115℃的沸點和1.20g/ml的密度。二乙基氯化鋁(deac)(97wt.%)購買自sigmaaldrich。deac具有125℃的沸點和0.961g/ml的密度。異丁基鋁氧烷(ibao)(2.7wt.%在庚烷中)購買自albemarle。其容納於玻璃瓶中,並且儲存在手套箱冷凍器內。ibao具有98℃的沸點和0.691g/ml的密度。三異丁基鋁(tibal)購買自akzonobel。tibal具有86℃的沸點和0.786g/ml的密度。乾燥劑(drieritetm)購買自sigmaaldrich。在使用之前,通過將乾燥劑在設置在260℃的馬弗爐中烘烤16小時的時間來對其進行調節。乾燥劑不含指示劑。2-氯-2-甲基丙烷(叔丁基氯或tbucl)購買自sigmaaldrich。通過在惰性環境下,以每100ml的tbucl30g乾燥劑的比率,將tbucl放置在經預乾燥的乾燥劑上約16小時,從而乾燥tbucl。在該處理過程中,將容納tbucl的燒瓶用箔遮蓋從而將其與光隔離,從而使異丁烯的形成最小化。通過真空轉移來將經乾燥的tbucl進一步純化。純化後,tbucl的水分含量為12ppm或更低,並具有大於97%的純度。在該流程中使用的全部玻璃器具在130℃的烘箱中乾燥過夜。乙烯按聚合級購買自praxair。通過使氣體經過一系列純化床來將乙烯純化和乾燥,所述純化床包括氧化鋁(品牌:selexsorbcos)、分子篩(類型:13x)和脫氧床(品牌:oxiclear®)。使購買的1-辛烯通過以1升批次儲存在3a分子篩上來乾燥。氯化鈦(iv)(ticl4)在氮氣下包裝按99.9%純度購買自sigmaaldrich。甲醇按gracs等級購買自emdchemicals。分析方法熔融指數(「mi」)測量根據astm方法d-1238來進行。聚合物密度使用astmd-1928來測量。聚合物分子量和分子量分布通過凝膠滲透色譜(gpc)來測量。儀器(waters150-c)在140℃下、在1,2,4-三氯苯中使用,並且使用聚乙烯標準物將其校準。聚合物支化頻率(polymerbranchfrequencies)通過ft-ir來確定。所使用的儀器為nicolet750magna-ir分光光度計。針對鈦價態分布對一些催化劑樣品進行分析。基於科學論文(j.c.w.chien等人,j.polym.sci.parta:polym.chem.1989,27,1499-1514)開發了用於鈦價態分布的氧化還原滴定法,並且基於astm標準e878-01開發了用於鈦含量分析的紫外(uv)法。電感耦合等離子體質譜(icp-ms)分析在agilent7700系列的儀器上完成。將樣品用5%的硝酸溶液消化,並在高能氦模式中分析從而除去任何的光譜幹擾。儀器使用經認證的標準物來校準。ti和mg標準物購買自spcsciences,並且cl購買自bdh。使用brukergeneralareadetectordiffractionsystem(gadds)來收集x射線衍射圖譜。x射線使用設置在30kv和30ma下的cu管(波長為1.54184a)來生成。樣品至檢測器距離為5.0cm。檢測器至樣品的角度(2θ)為30°。對於數據收集,將粉末化的樣品置於1.0mmid石英管中。對衍射圖譜進行背景校正。gpc-ft-ir:通過在烘箱中在150℃下將聚合物在1,2,4-三氯苯(tcb)中加熱並在轉輪(wheel)上旋轉4小時,從而製備聚合物樣品溶液(2至4mg/ml)。向混合物添加抗氧化劑2,6-二叔丁基-4-甲基苯酚(bht),從而使聚合物穩定化以免於氧化降解。bht濃度為250ppm。在watersgpc150c色譜裝置上,在140℃下對樣品溶液進行色譜分析,所述色譜裝置裝配有4個shodex柱(ht803、ht804、ht805和ht806),以1.0ml/分鐘的流量使用tcb作為流動相,其中ftir光譜儀和經由經加熱的傳遞管線而通過與色譜裝置耦合的單元(cell)的經加熱ftir流作為檢測系統。以250ppm的濃度向流動相添加bht,從而保護sec柱免於氧化降解。樣品注射體積為300ml。將原始ftir光譜用opusftir軟體進行處理,並且與opus關聯的chemometricsoftware(pls技術)來即時計算聚合物濃度和甲基含量。然後,獲得聚合物濃度和甲基含量,並且用cirrusgpc軟體對其進行基線校正。用窄分布的聚苯乙烯標準物來校準sec柱。使用mark-houwink公式將聚苯乙烯分子量換算為聚乙烯分子量,如在astm標準測試方法d6474中所描述的。基於通過如在由p.j.deslaurierspolymer2002,43,159-170出版的著作中描述的pls技術來預測的聚合物濃度和甲基含量,計算共聚單體含量。tref:將聚合物樣品(80至100mg)引入至polymerchar晶體-tref裝置的反應器容器中。用35ml1,2,4-三氯苯(tcb)填充反應器容器,將其加熱至所需的溶解溫度(例如150℃)2小時。然後,將溶液(1.5ml)載入填充有不鏽鋼珠的tref柱中。在給定的穩定化溫度(例如110oc)下使其能夠平衡45分鐘後,在溫度從穩定化溫度降至30℃(0.09℃/分鐘)的情況下使聚合物溶液能夠結晶。在30℃下平衡30分鐘後,用tcb(0.75ml/分鐘)、以30℃至穩定化溫度的溫度斜升(0.25℃/分鐘)洗脫結晶的樣品。在運轉結束時在溶解溫度下清潔tref柱30分鐘。使用polymerchar軟體、excel電子表格和自行開發的tref軟體來處理數據。cdbi定義為其組成在中值共聚單體組成(mediancomonomercomposition)的50%內的聚合物的百分比。其由組成分布曲線和組成分布曲線的標準化累計積分來計算,如在美國專利第5376439號中所例示的。epr是能夠檢測化學樣品中的未成對電子的磁共振技術。其通過觀察在用通常在微波頻率範圍內的單色輻射輻照時未成對電子共振所處於的磁場而發生。共振場的精確值對於電子的化學環境敏感,並且由g值來指定。g使用下式計算(atkins,peterw.,physicalchemistry,5thed.,1994,freemanpress,newyork):其中,h為普朗克常數(6.63×10-34j·s),ν為微波頻率(以hz計),µb為玻爾磁子(9.27×10-24j·t-1),並且b為磁場(以t計)。全部epr譜均在室溫下記錄於brukeremx10/12光譜儀上。場校準使用強間距(strongpitch)標準物來完成。將齊格勒納塔催化劑進行乾燥並填塞在4mm石英epr管中,並用環氧樹脂密封從而保持管內的惰性氛圍。epr模擬使用基於matlab的軟體包easyspin來進行。模擬的原理描述於論文(stefanstoll,arthurschweiger,「easyspin,acomprehensivesoftwarepackageforspectralsimulationandanalysisinepr」,j.magn.reson.2006,178,42-55)中。催化劑合成和表徵所有實驗均在手套箱中、在氮氣氛圍下使用油浴或者加熱罩作為熱源來進行。所使用的所有玻璃器具在鹼浴中清潔過夜,在酸浴中衝洗,用去離子水衝洗,然後放置在135℃的烘箱中過夜以乾燥。實施例1:mgcl2合成(δ-mgcl2)和表徵:在1000ml圓底燒瓶中,將16.910g(31.377mmol)的20.5wt.%bem添加至295ml癸烷。然後,在使用頂置式攪拌器以350rpm進行攪拌的同時,將溶液加熱至45℃的內溫(使用偶絲(thermowire)來監測)。將稀釋於5ml的癸烷中的7.149g(77.228mmol)的冷tbucl經由注射器一次性添加至bem溶液。將溶液在50℃下攪拌30分鐘。在形成mgcl2漿料後,將整個混合物轉移至500mlpyrex中。基於icp結果,cl/mg為2.33(mol/mol)。對於gadds結果,參見圖2。實施例2:催化劑1的合成:在1000ml圓底燒瓶中,將16.909g(30.304mmol)的19.8wt.%bem添加至295ml癸烷。然後,在使用頂置式攪拌器以350rpm進行攪拌的同時,將溶液加熱至45℃的內溫(使用偶絲來監測)。將稀釋於5ml癸烷中的7.072g(76.396mmol)冷tbucl經由注射器一次性添加至bem溶液。將溶液在50℃下攪拌30分鐘。在形成mgcl2後,在50℃下,使用注射劑將0.758g(3.996mmol)的ticl4添加至mgcl2。在添加ticl4之後,接著經由滴液漏鬥,以~3滴/秒的速率將稀釋於20ml癸烷中的6.229g(40.1871mmol)ibadc添加至反應。一旦完成添加衝洗漏鬥後,將漿料緩慢加熱至85℃的內溫,並將其攪拌1h。然後,經玻璃料(frit)過濾催化劑,用20ml癸烷洗滌1次,並用20ml環己烷洗滌4次,然後轉移至100ml玻璃pyrex瓶中,並在80ml的環己烷中再漿化。來自催化劑1的epr譜和通過模擬進行的光譜反卷積(spectraldeconvolution)實驗條件:頻率=9.389ghz,微波功率=12.7mw,時間常數=0.64ms,調製幅度=1g,8次42s的掃描的平均。模擬參數:a)g=[1.897、1.907、1.944],線寬=[90、170、60]高斯;b)g⊥=1.880,g||=1.945,線寬(⊥)=290高斯,線寬(||)=330高斯;c)g=1.950,線寬=50高斯;對信號強度的貢獻:a=18.6%,b=80.7%,c=0.7%。對於epr譜和模擬,參見圖4。實施例3:催化劑2a的合成:在1000ml圓底燒瓶中,將16.665g(30mmol)的19.9wt.%bem添加至295ml癸烷。然後,在使用頂置式攪拌器以350rpm進行攪拌的同時,將溶液加熱至45℃的內溫(使用偶絲來監測)。將稀釋於5ml癸烷中的6.942g冷tbucl經由注射器一次性添加至bem溶液。將溶液在50℃下攪拌30分鐘。在形成mgcl2後,在50℃下,使用漏鬥一次性將稀釋於5ml癸烷中的1.059g(6.8mmol)二乙基氯化鋁(ibadc)添加至mgcl2,並將溶液攪拌10分鐘。在50℃下使用注射器將用5ml癸烷稀釋的0.758g(4.0mmol)ticl4一次性全部添加至mgcl2。添加ticl4之後,經由移液管將稀釋於5ml癸烷中的3.430g(6.6mmol)的25wt.%二乙基乙醇鋁添加至反應。將漿料加熱至85℃的內溫,並且一旦溶液達到該溫度,就將溶液攪拌1h。然後,經玻璃料過濾催化劑,用20ml癸烷洗滌1次,並用20ml環己烷洗滌4次,並然後轉移至用於儲存的玻璃瓶中,並在80ml的環己烷中再漿化。實施例4:催化劑2b的合成:向3000ml圓底燒瓶,將107.792g(200mmol)的20.5wt.%bem添加至約520ml的來自冷凍器的冷癸烷。然後,在使用頂置式攪拌器以470rpm進行攪拌的同時,將溶液加熱至20℃的內溫(使用偶絲來監測)。將稀釋於70ml癸烷中的42.579g(460mmol)tbucl經由滴液漏鬥一次性添加至bem溶液。一旦完成添加衝洗漏鬥後,在將溶液加熱至50℃的同時,將溶液攪拌35分鐘。在形成mgcl2後,在50℃下使用滴液漏鬥將稀釋於30ml癸烷中的7.029g(45.3mmol)ibadc一次性添加至mgcl2。一旦完成添加衝洗漏鬥後,將溶液攪拌10分鐘。在添加ibadc之後,經由滴液漏鬥將稀釋於30ml癸烷中的5.057g(26.7mmol)ticl4一次性添加至反應。一旦完成添加衝洗漏鬥後,將漿料攪拌5分鐘。經由滴液漏鬥將稀釋於60ml癸烷中的22.904g(44mmol)deao一次性添加至反應。一旦完成添加衝洗漏鬥後,將溶液緩慢加熱至85℃的內溫,並將其攪拌1h。關閉加熱,並將溶液冷卻30分鐘。然後,經玻璃料過濾催化劑,用130ml癸烷洗滌1次,並用130ml環己烷洗滌2次。將固體催化劑轉移至用於儲存的玻璃瓶中,並用約350ml的環己烷再漿化。來自催化劑2b的epr譜和通過模擬進行的光譜反卷積。實驗條件:頻率=9.391ghz,微波功率=12.7mw,時間常數=0.64ms,調製幅度=1g,8次42s的掃描的平均。模擬參數:a)g=[1.899、1.899、1.949],線寬s=[120、120、120]高斯;b)g⊥=1.887,g||=1.945,線寬(⊥)=340高斯,線寬(||)=460高斯;c)g=1.950,線寬=42高斯;對信號強度的貢獻:a=22.3%,b=77.5%,c=0.2%。對於epr譜和模擬,參見圖5。實施例5:催化劑2c的合成:使用催化劑2b的流程,但在多批次合併的情況下製造催化劑。實施例6:催化劑3的合成:在1000ml圓底燒瓶中,將16.167g(30mmol)的20.5wt%bem添加至約285ml癸烷。然後,在使用頂置式攪拌器以345rpm進行攪拌的同時,將溶液加熱至45℃的內溫(使用偶絲來監測)。將稀釋於5ml癸烷中的6.387g(69mmol)tbucl經由滴液漏鬥一次性添加至bem溶液。一旦完成添加衝洗漏鬥後,在將漿料加熱至50℃的同時,將溶液攪拌30分鐘。在形成mgcl2後,在50℃下使用滴液漏鬥將稀釋於5ml癸烷中的4.231g(6.8mmol)的20.4wt.%etalcl2一次性添加至mgcl2。一旦完成添加衝洗漏鬥後,將溶液攪拌10分鐘。在添加etalcl2之後,經由滴液漏鬥將稀釋於5ml癸烷中的0.761g(4.0mmol)的ticl4一次性添加至反應。一旦完成添加衝洗漏鬥後,將漿料攪拌5分鐘。經由滴液漏鬥將稀釋於10ml癸烷中的3.434g(6.6mmol)deao一次性添加至反應。一旦完成添加衝洗漏鬥後,將溶液緩慢加熱至85℃的內溫,並將其攪拌1h。關閉加熱,並將溶液冷卻30分鐘。然後,經玻璃料過濾催化劑,用20ml癸烷洗滌1次,並用20ml環己烷洗滌2次。將固體催化劑轉移至用於儲存的玻璃瓶中,並用約80ml的環己烷再漿化。通過icp對催化劑的wt%ti進行分析,並確定為4.06。實施例7:催化劑4的合成:在1000ml圓底燒瓶中,將16.167g(30mmol)的20.5wt%bem添加至約285ml癸烷。然後,在使用頂置式攪拌器以350rpm進行攪拌的同時,將溶液加熱至45℃的內溫(使用偶絲來監測)。將稀釋於5ml癸烷中的6.387g(69mmol)tbucl經由滴液漏鬥一次性添加至bem溶液。一旦完成添加衝洗漏鬥後,在將漿料加熱至50℃的同時,將漿料攪拌30分鐘。在形成mgcl2後,在50℃下使用滴液漏鬥將稀釋於5ml癸烷中的0.825g(6.8mmol)二乙基氯化鋁(deac)一次性添加至mgcl2。一旦完成添加衝洗漏鬥後,將漿料攪拌10分鐘。在添加deac之後,經由滴液漏鬥將稀釋於5ml癸烷中的0.755g(4.0mmol)ticl4一次性添加至反應。一旦完成添加衝洗漏鬥後,通過漏鬥全部一次性添加用5ml癸烷稀釋的1.215g(2.33mmol)的25wt%deao,並且將漿料加熱至85℃的內溫,並將溶液攪拌1h。關閉加熱,並將溶液冷卻30分鐘。然後,經玻璃料過濾催化劑,用50ml癸烷洗滌1次,並用20ml環己烷洗滌2次。將固體催化劑轉移至用於儲存的玻璃瓶中,並用約80ml的環己烷再漿化。通過icp對催化劑的wt%ti進行分析,並確定為3.905。實施例8:催化劑5的合成:向1000ml圓底燒瓶,將16.167g(30mmol)的20.5wt.%bem添加至約285ml癸烷。然後,在使用頂置式攪拌器以350rpm進行攪拌的同時,將溶液加熱至45℃的內溫(使用偶絲來監測)。將稀釋於5ml癸烷中的6.395g(69mmol)tbucl經由滴液漏鬥一次性添加至bem溶液。一旦完成添加衝洗漏鬥後,在將漿料加熱至50℃的同時,將漿料攪拌30分鐘。在形成mgcl2後,在50℃下使用滴液漏鬥將稀釋於5ml癸烷中的1.052g(6.8mmol)ibadc一次性添加至mgcl2。一旦完成添加衝洗漏鬥後,將漿料攪拌10分鐘。在添加ibadc之後,經由滴液漏鬥將稀釋於5ml癸烷中的0.761g(4.0mmol)ticl4一次性添加至反應。一旦完成添加衝洗漏鬥後,將漿料加熱至85℃的內溫。經約20分鐘逐滴緩慢添加用癸烷稀釋的10.054g(2.10mmol)的2.7wt%ibao。然後,將溶液攪拌1h。關閉加熱,並將漿料冷卻30分鐘。然後,經玻璃料過濾催化劑,用20ml癸烷洗滌1次,並用20ml環己烷洗滌2次。將固體催化劑轉移至用於儲存的玻璃瓶中,並用約80ml的環己烷再漿化。通過icp對催化劑的wt%ti進行分析,並確定為3.33。實施例9:催化劑6的合成:向1000ml圓底燒瓶,將16.172g(30mmol)的20.5wt.%bem添加至約285ml癸烷。然後,在使用頂置式攪拌器以350rpm進行攪拌的同時,將溶液加熱至45℃的內溫(使用偶絲來監測)。將稀釋於5ml癸烷中的6.389g(69mmol)tbucl經由滴液漏鬥一次性添加至bem溶液。一旦完成添加衝洗漏鬥後,在將漿料加熱至50℃的同時,將漿料攪拌30分鐘。在形成mgcl2後,在50℃下使用滴液漏鬥將稀釋於5ml癸烷中的1.060g(6.8mmol)ibadc一次性添加至mgcl2。一旦完成添加衝洗漏鬥後,將漿料攪拌10分鐘。在添加ibadc之後,經由滴液漏鬥將稀釋於5ml癸烷中的0.760g(4.0mmol)ticl4一次性添加至反應。一旦完成添加衝洗漏鬥後,將漿料加熱至85℃的內溫。經約60分鐘,逐滴緩慢添加用癸烷稀釋的3.189g(4.10mmol)的25.7wt%tibal。然後,將溶液攪拌1h。關閉加熱,並將溶液冷卻30分鐘。然後,經玻璃料過濾催化劑,用20ml癸烷洗滌1次,並用20ml環己烷洗滌2次。將固體催化劑轉移至用於儲存的玻璃瓶中,並用約80ml的環己烷再漿化。對比實施例1:催化劑7的合成:在1000ml圓底燒瓶中,將16.665g(30mmol)的19.9wt.%bem添加至約285ml癸烷。然後,在使用頂置式攪拌器以350rpm進行攪拌的同時,將溶液加熱至45℃的內溫(使用偶絲來監測)。將稀釋於5ml癸烷中的6.942g(75mmol)冷tbucl經由注射器一次性添加至bem溶液。在將漿料加熱至50℃的同時,將漿料攪拌30分鐘。在形成mgcl2後,在50℃下使用注射器將稀釋於5ml癸烷中的0.758g的ticl4一次性全部添加至mgcl2。在添加ticl4之後,在將漿料加熱至85℃的同時,經由移液管將稀釋於5ml癸烷中的5.880g(11.3mmol)25wt.%二乙基乙醇鋁添加至反應(每次1-2ml)。在每次添加之間,將漿料攪拌15分鐘,並且溫度緩慢升高至85℃(即,在50、60、70℃下添加直至顏色不再變化)。當顏色不再變化時,停止添加。一旦其達到溫度,將漿料攪拌1h。然後,經玻璃料過濾催化劑,用20ml癸烷洗滌1次,並用20ml環己烷洗滌4次。然後,將固體催化劑轉移至用於儲存的玻璃瓶中,並用約80ml的環己烷再漿化。對比實施例2:催化劑8的合成:在母液去除而無洗滌步驟的情況下,完全按催化劑2製造催化劑。基於us7,666,810b2中的公開內容的對比實施例a在手套箱內,在1000ml圓底燒瓶中,將16.665g(30mmol)的19.9wt.%bem添加至100ml環己烷。然後,將溶液進一步用另外的190ml環己烷稀釋,並置於油浴中。燒瓶裝配有含熱偶絲的冷凝器、漿式攪拌器和間隔物。使用頂置式攪拌器,將溶液以400rpm攪拌並加熱至45℃。經由氣密注射器,一次性添加稀釋於約5ml環己烷中的5.95ml(54mmol)tbucl。將溶液在50℃下攪拌半小時。過濾催化劑漿料並用環己烷洗滌3次(每次20ml)。使用注射器,以mg:ti=7.5的比率,在50℃下將1.79ml的2.24mticl4溶液添加至astmgcl2。將溶液攪拌半小時,然後過濾並用環己烷洗滌3次(每次20ml)。將催化劑在70ml的環己烷中再漿化,並轉移至用於儲存的玻璃瓶中。將少量的催化劑漿料乾燥,並製備固體樣品用於epr分析。來自製備對比實施例a的epr譜和通過模擬進行的光譜反卷積:實驗條件:頻率=9.395ghz,微波功率=12.7mw,時間常數=0.64ms,調製幅度=1g,8次42s的掃描的平均。模擬參數:a)g=[1.910、1.898、1.955],線寬s=[175、115、75]高斯;b)g⊥=1.883,g||=1.972,線寬(⊥)=235高斯,線寬(||)=200高斯;c)g=1.969,線寬=37高斯;對信號強度的貢獻:a=52.4%,b=47.3%,c=0.3%。對於epr譜和模擬,參見圖6。表1催化劑特性在實驗室規模的連續式聚合裝置中測試離線(offline)催化劑連續式聚合在連續式聚合裝置(cpu)上進行。cpu包含71.5ml攪拌反應器,並且對聚合實驗而言,在160至280℃運行。具有20ml體積的上遊混合反應器在比聚合反應器低5℃的溫度下運行。使用混合反應器來預熱乙烯、辛烯和一些溶劑物流。以連續過程直接向聚合反應器添加催化劑進料和剩餘的溶劑。保持27ml/分鐘的進入聚合反應器的總連續流量。在漿料遞送系統中,將來自上述實施例的催化劑添加至cpu。漿料遞送系統由帶有3500ml攪拌漿料儲存器的倒置1000ml注射泵構成。漿料通過壓差從攪拌瓶經由不鏽鋼套管而轉移至3500ml攪拌漿料儲存器中。然後,在儲存器中將漿料用經純化的環己烷稀釋至所需濃度。一旦將漿料轉移並稀釋,在將任何漿料轉移至注射泵中之前,將其在儲存器中攪拌最少15分鐘。當漿料就緒以轉移至反應器時,打開氣動電磁閥以使得漿料流動至注射器筒,所述氣動電磁閥將儲存器與注射器筒相隔離。然後,在注射器筒中恆定攪拌的情況下,以25ml/分鐘的流量將注射器筒裝載至期望的體積。當將注射器筒填充至所需體積時,關閉至儲存器的電磁閥,將注射器筒和電磁閥相隔離。然後,在仍與反應器相隔離的同時,使注射器筒達到反應器壓力。當注射器筒已達到反應器壓力時,打開氣動電磁閥(其將注射器筒與反應器相隔離)。然後,校準注射泵,並進行程序控制從而遞送所需的漿料流量。對於漿料催化劑實驗,以0.5的辛烯/乙烯重量比來製造共聚物。在聚合反應器中,以10wt.%的乙烯濃度進料乙烯。cpu系統在10.5mpa的壓力下運行。在進入反應器之前,溶劑、單體和共聚單體物流均通過cpu系統純化。q是乙烯轉化率(如通過在線氣相色譜(gc)而確定),並且聚合活性kp定義如下:(kp)(hut)=q((1-q)(1/催化劑濃度)其中,q為所轉化的乙烯單體的分數;hut是聚合反應器中的倒易空間速度(保持時間),以分鐘表達,並且貫穿整個實驗程序保持恆定;並且催化劑濃度為聚合反應器中的濃度,以mmolti/l表達,並且漿料催化劑的ti濃度通過icp確定。所有聚合實驗在220℃下進行,並且以90±1的乙烯轉化率和2至4的二乙基乙醇鋁(deao)與ti的摩爾比收集聚合物。表2cpu上的催化劑性能運行#催化劑代號cj/ti比率乙烯轉化率qkp(1/mm*分鐘)mw(10-3)pdbr/1000c1催化劑13.690.259.066.12.710.22催化劑2a2.089.789.857.43.012.43催化劑2b3.390.562.573.63.510.54催化劑2c2.289.771.768.73.011.15催化劑32.290.669.370.13.110.96催化劑42.190.674.966.62.610.07催化劑52.390.068.967.92.910.98催化劑62.089.668.878.42.710.79催化劑71.590.457.165.13.39.710催化劑82.189.972.961.83.111.7對以試驗性工廠(pilotplant)規模的連續聚合設施進行的本發明的離線齊格勒納塔(z/n)漿料催化劑(催化劑2c)和作為對比實施例3的通過管線內形成的zn催化劑製造的催化劑9的測試。表3中的實施例例示了使用單個試驗性工廠反應器系統和離線漿料催化劑(催化劑2c)在中壓下的乙烯和1-辛烯的連續流溶液共聚合。第一反應器為具有24.0升體積的連續攪拌釜式反應器(cstr)。第二反應器為具有cstr體積的10%的體積(2.4升)的管式反應器。如表3中說明地,向cstr中進料單體、溶劑和催化劑(操作模式1)。在實驗中,使用具有由二乙基乙醇鋁(deao)或三異丁基鋁(tibal)構成的活化劑的離線齊格勒納塔漿料催化劑(催化劑2c)。為了比較本發明的催化劑2c,使用對比的管線內形成的齊格勒納塔(z/n)催化劑體系(催化劑9),並且在下一部分對其進行描述。使用漿料遞送系統,將催化劑2c泵入連續流聚合反應器。漿料遞送系統由漿料筒、攪動漿料日量槽、再循環迴路、漿料催化劑計量泵、和溶劑稀釋劑迴路構成。通過用氮氣對筒進行加壓/鼓泡,以數次裝料將經稀釋的漿料催化劑從漿料筒轉移至漿料日量槽。一旦將漿料催化劑轉移至漿料催化劑日量槽中,啟動攪動器和再循環泵以將催化劑漿料保持懸浮和恆定組成。將經稀釋的漿料催化劑的溫度保持在環境溫度。將槽壓力保持在300kpag。當漿料催化劑就緒以轉移至反應器時,啟動漿料催化劑遞送泵,並且將漿料催化劑集中至泵。在漿料催化劑遞送泵的排出口處,使用高流動溶劑(highflowsolvent)稀釋劑從而使漿料催化劑保持懸浮,並且輔助催化劑向反應器的遞送。將稀釋劑流量保持在15kg/hr。將溶劑的溫度控制在25℃。將溶劑和漿料催化劑泵送至流量傳感器,並且記錄流量。進入反應器的漿料催化劑流量通過稀釋劑流量與合併的稀釋劑和漿料催化劑的流量之間的差來計算。進入反應器的漿料催化劑流量(和ppms)通過改變漿料催化劑遞送泵的馬達變頻驅動、或者泵推送器(pumpstroker)來調節。催化劑流量具有如在表中示出的表達為ti的按重量計的百萬分率的目標設定值,並且對其進行調節以將總乙烯轉化率保持為大於80%。使用如上提及的管線內形成的齊格勒納塔催化劑體系(催化劑9),其由四氯化鈦(ticl4)、丁基乙基鎂(bem)和叔丁基氯(tbucl)構成,具有由三乙基鋁(teal)或二乙基乙醇鋁(deao)構成的活化劑。bem和teal以「預混合」(20/1mg/al摩爾比)的形式提供。在catalysttorpedo內,將所有催化劑組分混合於甲基戊烷溶劑中。混合順序為bem/teal和tbucl(段#1);接著ticl4(段#2);然後接著deao(段#3)。將催化劑與甲基戊烷溶劑一同泵送至反應器中。催化劑流量具有表達為ti的按重量計的百萬分率的目標設定值,並且對其進行調節以將總乙烯轉化率保持為大於80%。因此,在表3中,對於給定的熔融指數、密度和應力指數,產物1、即在該反應器配置中製備的產物建立了「基線」反應器操作條件。產物2使用具有相同的鋁活化劑的催化劑2c來製造。將反應器操作條件調節至生成與產物3中相似的熔融指數、密度和應力指數,產物3使用三異丁基鋁(tibal)作為活化劑來製備。表4中的實施例例示了使用單個試驗性工廠反應器系統和離線漿料催化劑(催化劑2c)在中壓下的乙烯和1-辛烯的連續流溶液共聚合。第一反應器為具有24.0升體積的連續攪拌釜式反應器(cstr)。第二反應器為具有cstr體積的82%的體積(19.7升)的管式反應器。將催化劑進料至cstr中。如表4中說明地,將單體和溶劑在兩個反應器之間拆分(操作模式2)。為了對比,使用管線內形成的齊格勒納塔催化劑體系(催化劑9)。因此,在表4中,對於給定的熔融指數、密度和應力指數,產物4、即在該反應器配置中製備的產物建立了「基線」反應器操作條件。產物5使用具有相同的活化劑的離線漿料催化劑(催化劑2c)來製造。將反應器操作條件調節至生成與產物4中相似的熔融指數、密度和應力指數。產物6使用三異丁基鋁(tibal)作為活化劑而生成。在表中使用的其他縮寫的列表如下:hr:小時conc:濃度wt%:重量百分比wt/wt:重量/重量temp:溫度c:攝氏度rpm:轉/分鐘mol:摩爾或摩爾的ppm:按重量計的百萬分率。表3在操作模式1下的催化劑2c和催化劑9的性能表4在操作模式2下的催化劑2c和催化劑9的性能聚合物特性通過gpc-ft-ir和針對cdbi的tref進一步對產物5進行表徵。gpc-ft-ir示出相對平坦的共聚物引入,並且來自催化劑2c的產物5的cdbi為58.4。由產物2、3、5和6以及來自對比實施例3和4的產物1和4來製備膜。在常規的吹塑生產線上製造所述膜,其通過具有2.5英寸螺杆直徑的單螺杆擠出機來進料。通過電動馬達來驅動擠出機。向全部擠出操作添加常規的添加劑(抗氧化劑和加工助劑)。迫使擠出物穿過具有4英寸直徑和35mil模具間隙的圓形模具。使用2.5:1的吹脹比(bur)從而以100lbs/hr的輸出率製備膜。對於1mil膜,產物2、3、5和6以及對比實施例3和4中的產物1和4的膜特性(例如落鏢衝擊、1%割線模量、md撕裂、td撕裂、霧度和己烷可提取性)基本上相同(在實驗誤差內)。工業實用性本發明涉及用於聚烯烴的高溫溶液聚合的鎂-鈦催化劑。當前第1頁12