一種雙烷氧基硫代膦酸酯的製備方法與流程
2023-10-05 21:04:29 4
本發明屬於有機合成技術領域,特別涉及一種雙烷氧基硫代膦酸酯的製備方法。
背景技術:
雙烷氧基硫代膦酸酯是一類在有機合成中重要的中間體,在醫藥、農藥和顏料等具有重要的應用價值。現有技術已公開了若干種合成雙烷氧基硫代膦酸酯的方法,但這些方法存在著以下問題:原料味道大、反應條件苛刻、鹼用量大等。
其中,一篇cn104292255介紹了「一種s-芳基硫代磷酸酯的製備方法」,公開了以芳基磺醯氯和亞磷酸酯為原料,銅鹽的催化下,在60~150℃反應12~24h,製得目標產物。該方法反應溫度高,在實施例製備的化合物均需要在120℃以上,且反應時間長,需24h。因此,尋找一種溫和的方法合成雙烷氧基硫代膦酸酯成為有機合成中亟待解決的難題。
技術實現要素:
本發明提供一種雙烷氧基硫代膦酸酯的製備方法,以芳基磺醯氯和亞磷酸二烷基酯為原料,在銅鹽催化和l-脯氨酸作配體下,在80~100℃下反應10~20h,製得雙烷氧基硫代膦酸酯。
本發明的技術方案如下:
一種雙烷氧基硫代膦酸酯的製備方法,其特徵在於,包括以下步驟:
(1)將芳基磺醯氯和亞磷酸二烷基酯加入到有機溶劑中,再加入銅鹽催化劑和l-脯氨酸配體,在80~100℃下反應10~20h;
(2)反應結束後,濃縮反應液,經分離純化後得到雙烷氧基硫代膦酸酯。
優選為,所述的芳基磺醯氯和亞磷酸二烷基酯的摩爾比是1:2-1:4。
優選為,每摩爾所述的芳基磺醯氯加入2-10l有機溶劑。
優選為,所述的芳基磺醯氯取代有烷氧基、烷基、滷素或硝基中的一種。
優選為,所述的有機溶劑為四氫呋喃。
優選為,所述的銅鹽催化劑為二水合氯化銅。
優選為,所述的銅鹽催化劑的摩爾百分數為5-10%.
優選為,所述的l-脯氨酸配體的摩爾百分數為10-20%。
優選為,步驟(2)分離純化時,濃縮得到的反應物以乙酸乙酯/石油醚=1/3-1/10(v/v)為展開劑進行柱層析分離。
與現有技術相比,本發明的有益效果如下:
一、本發明的一種雙烷氧基硫代膦酸酯的製備方法,以價廉易得的芳基磺醯氯和亞磷酸二烷基酯為原料,無需鹼性環境,僅在催化量的銅鹽和配體下,在溶液中即可發生偶聯反應,製備得到雙烷氧基硫代膦酸酯類化合物;
二、本發明的製備方法,底物適用範圍廣,對烷氧基、烷基、滷素、硝基等多種基團取代的芳基磺醯氯,具有較好的適用性;且本發明的製備方法工藝簡單,操作方便,底物範圍廣,具有較優的產率,適合推廣應用,製備的產品可用於醫藥、農藥等眾多領域。
附圖說明
圖1為實施例1的o,o-二乙基-s-(4-甲基苯基)硫代膦酸酯1hnmr氫譜圖;
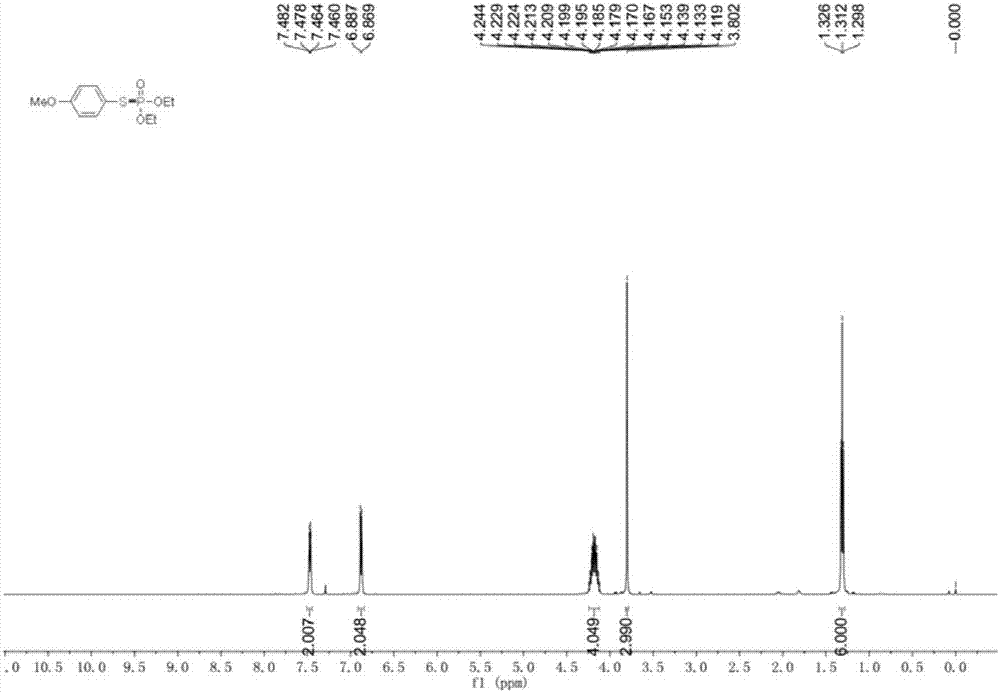
圖2為實施例2的o,o-二乙基-s-(4-甲氧基苯基)硫代膦酸酯1hnmr氫譜圖;
圖3為實施例3的o,o-二乙基-s-(3-硝基苯基)硫代膦酸酯1hnmr氫譜圖。
具體實施方式
下面結合具體實施例,進一步闡述本發明。應該理解,這些實施例僅用於說明本發明,而不用於限定本發明的保護範圍。在實際應用中本領域技術人員根據本發明做出的改進和調整,仍屬於本發明的保護範圍。
本發明的一種雙烷氧基硫代膦酸酯的製備方法,具體包括以下步驟:
(1)將芳基磺醯氯和亞磷酸二烷基酯加入到四氫呋喃中,再加入二水合氯化銅催化劑和l-脯氨酸配體,在80~100℃下反應10~20h;
(2)反應結束後,濃縮反應液,經分離純化後得到雙烷氧基硫代膦酸酯。
本發明涉及的反應通式如下:
其中,r1為甲基、乙基、異丙基的一種或多種;r2為未取代或者帶有取代基的苯基,所述取代基為烷氧基、硝基、烷基或滷素中的一種。
實施例1
o,o-二乙基-s-(4-甲基苯基)硫代膦酸酯的製備方法,包括下列步驟:
稱取2.0mmol的亞磷酸二乙酯與0.5mmol對甲基苯磺醯氯於反應瓶中,加入5%的二水合氯化銅作為催化劑,10%的l-脯氨酸作為配體,加入1.0ml四氫呋喃,80℃下反應12h。反應液減壓濃縮後,以乙酸乙酯/石油醚=1:3(v/v)為展開劑柱層析分離,得98mg目標化合物。
本實施例1的目標產品收率為75%。
如圖1所示,對目標產品進行核磁表徵,如下:1hnmr(500mhz,cdcl3):δ7.44(d,j=7.0hz,2h),7.15(d,j=8.0hz,2h),4.25-4.12(m,4h),2.34(s,3h),1.31(t,j=7.0hz,3h).
實施例2
o,o-二乙基-s-(4-甲氧基苯基)硫代膦酸酯的製備方法,包括下列步驟:
稱取2.0mmol的亞磷酸二乙酯與0.5mmol對甲氧基苯磺醯氯於反應瓶中,加入5%的二水合氯化銅作為催化劑,10%的l-脯氨酸作為配體,加入1.0ml四氫呋喃,80℃下反應12h。反應液減壓濃縮後,以乙酸乙酯/石油醚=1:3(v/v)為展開劑柱層析分離,得94mg目標化合物。
本實施例2的目標產品收率為68%。
如圖2所示,對目標產品進行核磁表徵,如下:1hnmr(500mhz,cdcl3):δ7.48-7.46(m,2h),6.88(d,j=9.0hz,2h),4.24-4.12(m,4h),3.80(s,3h),1.31(t,j=7.0hz,6h).
實施例3
o,o-二乙基-s-(3-硝基苯基)硫代膦酸酯的製備方法,包括下列步驟:
稱取2.0mmol的亞磷酸二乙酯與0.5mmol3-硝基苯磺醯氯於反應瓶中,加入5%的二水合氯化銅作為催化劑,10%的l-脯氨酸作為配體,加入1.0ml四氫呋喃,80℃下反應12h。反應液減壓濃縮後,以乙酸乙酯/石油醚=1:3(v/v)為展開劑柱層析分離,得101mg目標化合物。
本實施例的目標產品收率為69%。
如圖3所示,對目標產品進行核磁表徵,如下:1hnmr(500mhz,cdcl3):δ8.45-8.44(m,1h),8.23(d,j=8.0hz,1h),7.94(d,j=8.0hz,1h),7.57(t,j=8.0hz,1h),4.31-4.19(m,4h),1.36(t,j=7.0hz,6h).
實施例4
o,o-二乙基-s-(2,4,6-三甲基苯基)硫代膦酸酯的製備方法,包括下列步驟:
稱取2.0mmol的亞磷酸二乙酯與0.5mmol2,4,6-三甲基苯磺醯氯於反應瓶中,加入5%的二水合氯化銅作為催化劑,10%的l-脯氨酸作為配體,加入1.0ml四氫呋喃,80℃下反應12h。反應液減壓濃縮後,以乙酸乙酯/石油醚=1:3(v/v)為展開劑柱層析分離,得93mg目標化合物。
本實施例4的目標產品收率為64%。
對目標產品進行核磁表徵,如下:1hnmr(500mhz,cdcl3):δ6.94(s,2h),4.17-4.05(m,4h),2.53(s,6h),2.25(d,j=2.0hz,3h),1.28(t,j=7.0hz,6h).
實施例5
o,o-二甲基-s-(4-氯苯基)硫代膦酸酯的製備方法,包括下列步驟:
稱取2.0mmol的亞磷酸二甲酯與0.5mmol對氯苯磺醯氯於反應瓶中,加入5%的二水合氯化銅作為催化劑,10%的l-脯氨酸作為配體,加入1.0ml四氫呋喃,80℃下反應12h。反應液減壓濃縮後,以乙酸乙酯/石油醚=1:3(v/v)為展開劑柱層析分離,得76mg目標化合物。
本實施例5的目標產品收率為60%。
對目標產品進行核磁表徵,如下:1hnmr(500mhz,cdcl3):δ7.50(d,j=7.0hz,2h),7.33(d,j=8.5hz,2h),3.84(s,3h),3.81(s,3h).
以上公開的本發明優選實施例只是用於幫助闡述本發明。優選實施例並沒有詳盡敘述所有的細節,也不限制該發明僅為所述的具體實施方式。顯然,根據本說明書的內容,可作很多的修改和變化。本說明書選取並具體描述這些實施例,是為了更好地解釋本發明的原理和實際應用,從而使所屬技術領域技術人員能很好地理解和利用本發明。本發明僅受權利要求書及其全部範圍和等效物的限制。