可逆雙羥基菲並咪唑Hg2+螢光探針合成與使用方法與流程
2023-12-10 05:17:31 4
本發明涉及一種Hg2+可逆螢光探針及其合成與使用方法。
背景技術:
Hg2+是一種極具生理毒性的重金屬離子,具有持久性、易遷移性和高度的生物富集性,是目前全球最引人關注的環境汙染物之一。由於對動植物和人體健康都有很大的影響,比如對人體的腦、腎臟及內分泌系統造成巨大損害。因此對汞離子的檢測具有非常重要的意義。近年來,越來越多科研工作者開始了離子螢光探針的研究工作。徐鑑報導了一種新型耐爾藍衍生物對Hg2+的識別性能[化學試劑 (2015, 37 ( 2), 158 ~ 160; 164) ]。研究發現Hg2+可以選擇性淬滅這種耐爾藍衍生物的螢光;2016年,《高等學校化學學報》報導了具有高選擇性Hg2+比率螢光探針 [高等學校化學學報 (2016, 37 (2), 232 ~ 238) ]。在日光下,Hg2+可以使主體溶液的顏色由淺綠色變為橙色。2010年在《四面體》( Tetrahedron ) 的66期的3662-3667頁公開的文章A new bis(pyrenyl) azadiene-based probe for the colorimetric and fluorescent sensing of Cu(II) and Hg(II) 和2012年在《有機&生物化學》( Organic &Biomolecular Chemistry) 的2733-2738頁公開的文章 Alteration of selectivity in rhodamine based probes for Fe(III) and Hg(II) ion induced dual mode signalling responses 都是關於汞離子識別的技術。這兩篇文章中,主體化合物除了可以識別Hg2+外,還分別對Cu(II)和Fe(III)具有識別性能。
根據目前文獻報導的對於Hg2+螢光探針的研究,主要存在以下兩個缺陷:
1、Hg2+對主體化合物的螢光只能進行萃滅或增強的一次性的識別性能,而沒有可逆識別性能;
2、其他金屬離子與Hg2+共存時,主體化合物除了識別Hg2+,還對其他金屬離子有識別性能,所以,識別不具有專一性。
技術實現要素:
本發明是要解決現有的其他金屬離子對Hg2+的識別存在幹擾以及缺少可逆識別性能的技術問題,而提供的主體化合物對Hg2+具有專一識別性能並實現可逆(off-on-off)螢光信號響應檢測的可逆雙羥基菲並咪唑Hg2+螢光探針及其合成與使用方法。
本發明的可逆雙羥基菲並咪唑Hg2+螢光探針的結構式為:
上述的可逆雙羥基菲並咪唑Hg2+螢光探針的製備方法,按以下步驟進行:
一、中間體化合物的合成:
將菲醌、5-硝基水楊醛和乙酸銨按照1 : 1.5 : 2的摩爾比投入到反應器中,再加入冰乙酸作為溶劑,升溫至80 ~ 110℃並攪拌6 ~ 12 h後,反應停止,冷卻至室溫,向反應器中加入水,再用10%的氫氧化鈉溶液調pH值至8 ~ 10,抽濾,得到黃色固體。乾燥後,再用乙酸乙酯重結晶,再抽濾、乾燥,得到中間體化合物;
二、中間體化合物的合成:
稱取2.0 mmol中間體化合物和 0.35 g 雷尼鎳加入到反應器中,再加入乙醇作為溶劑,通入氮氣保護,在攪拌條件下,滴加7.5 mL質量百分濃度為80%的水合肼溶液,滴加完畢後,升溫至50 ~ 80℃,反應5 ~ 10 h,冷卻至室溫,抽濾,用乙酸乙酯洗滌,旋轉蒸發去除濾液後,得到褐色固體並乾燥。再用乙酸乙酯重結晶,再抽濾、乾燥,得到中間體化合物;
三、Hg2+螢光探針的合成:
按照1 : 1 ~ 3的摩爾比稱取中間體化合物和水楊醛,加入到反應器中,再向其中加入酸性介質作為溶劑,常溫攪拌反應1 ~ 5 h。反應結束後,向反應體系中加入水淬滅反應,再用10%氫氧化鈉溶液調pH值至8 ~ 10,析出固體。然後進行抽濾,濾餅用水洗至中性,乾燥,再用乙酸乙酯重結晶,得到可逆雙羥基菲並咪唑Hg2+螢光探針MQ。
步驟三中所述的酸性介質進一步優選為質量百分濃度為98%的濃硫酸、質量百分濃度為65 ~ 68%的濃硝酸、冰乙酸或苯甲酸。
本發明的合成過程可用下面的式子表示:
本發明提供了一種在水相體系中,pH為6.5 ~ 7.4的環境內,具有高選擇性、高靈敏性且不受其他金屬離子幹擾的可逆雙羥基菲並咪唑Hg2+螢光探針。該螢光探針能選擇性識別Hg2+,不受K+、Ba2+、Ca2+、Na+、Mg2+、Zn2+、Cr3+、Cd2+、Ni2+、Co2+、Pb2+、Cu2+、Ag+、Al3+ 和Fe3+等其他金屬離子的幹擾,螢光分子-Hg2+絡合物可以用來對S2–進行識別檢驗,完成該螢光分子探針的可逆(off-on-off)螢光信號響應,不受其他陰離子AcO–、Cl–、H2PO4–、HCO3–、CO32–、HPO42–、HSO3–、NO2–、Br–、SO32–、SO42–、BrO3–和PO43–的幹擾。
附圖說明
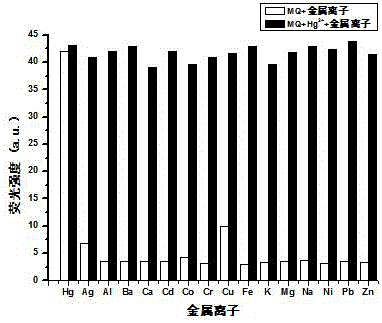
圖1是試驗1製備的可逆雙羥基菲並咪唑Hg2+螢光探針對不同金屬離子的螢光發射光譜圖。
圖2是試驗1製備的可逆雙羥基菲並咪唑Hg2+螢光探針在其他金屬離子存在的情況下,Hg2+螢光探針的螢光發射光譜圖。
圖3是試驗1製備的該可逆螢光分子-Hg2+絡合物對不同的陰離子的螢光發射光譜圖。
圖4是試驗1製備的該可逆螢光分子-Hg2+絡合物在其他陰離子存在的情況下,S2–螢光探針的螢光發射光譜圖。
具體實施方式
具體實施方式一:本實施方式的可逆雙羥基菲並咪唑Hg2+螢光探針的結構式為:
具體實施方式二:具體實施方式一的可逆雙羥基菲並咪唑Hg2+螢光探針的製備方法,按以下步驟進行:
一、中間體化合物的合成:
將菲醌、5-硝基水楊醛和乙酸銨按照1 : 1.5 : 2摩爾比加入到反應器中,再加入冰乙酸作為溶劑,升溫至80 ~ 110℃並攪拌6 ~ 12 h後,冷卻至室溫,向反應器中加入水,再用10%的氫氧化鈉溶液調pH值至8 ~ 10,抽濾,得到黃色固體。乾燥後,再用乙酸乙酯重結晶,再抽濾、乾燥,得到中間體化合物;
二、中間體化合物的合成:
稱取2.0 mmol中間體化合物和 0.35 g 雷尼鎳加入到反應器中,再加入乙醇作為溶劑,通入氮氣保護,在攪拌條件下,滴加7.5 mL質量百分濃度為80%的水合肼溶液,滴加完畢後,升溫至50 ~ 80℃,反應5 ~ 10 h,冷卻至室溫,抽濾,用乙酸乙酯洗滌,旋轉蒸發去除濾液後,得到褐色固體並烘乾。再用乙酸乙酯重結晶,再抽濾、乾燥,得到中間體化合物;
三、Hg2+螢光探針的合成:
按照1 : 1 ~ 3的摩爾比稱取中間體化合物和水楊醛,加入到反應器中,再加入酸性介質為溶劑,常溫攪拌反應1 ~ 5 h,反應結束後,向反應體系中加入水淬滅反應,再用10%氫氧化鈉溶液調pH值至8 ~ 10,析出固體,然後進行抽濾,濾餅用水洗至中性,乾燥,再用乙酸乙酯重結晶,得到可逆雙羥基菲並咪唑Hg2+螢光探針。
具體實施方式三:本實施方式與具體實施方式二不同的是步驟三中所述的酸性介質為質量百分濃度為98%的濃硫酸、質量百分濃度為65 ~ 68%的濃硝酸、冰乙酸或苯甲酸。
其它與具體實施方式二相同。
具體實施方式四:本實施方式與具體實施方式二或三不同的是步驟一中的反應溫度為100℃,反應時間為10 h。其它與具體實施方式二或三相同。
具體實施方式五:本實施方式與具體實施方式二至四之一不同的是步驟一中的pH值至9。其它與具體實施方式二至四之一相同。
具體實施方式六:本實施方式與具體實施方式二至五之一不同的是步驟二中反應溫度為70℃,反應時間為8 h。其它與具體實施方式二至五之一相同。
具體實施方式七:本實施方式與具體實施方式二至六之一不同的是步驟三中常溫攪拌反應3 h。其它與具體實施方式二至六之一相同。
具體實施方式八:本實施方式與具體實施方式二至七之一不同的是步驟三中pH值為9。其它與具體實施方式二至七之一相同。
具體實施方式九:本實施方式與具體實施方式二至八之一不同的是步驟三中反應物的摩爾比為1 : 3。
用以下實例驗證本發明的有益效果:
試驗1:本試驗的可逆雙羥基菲並咪唑Hg2+螢光探針的製備方法,按以下步驟進行:
一、中間體化合物的合成:
稱取菲醌5 mmol、5-硝基水楊醛7.5 mmol、乙酸銨10 mmol加入到50 mL的三口瓶中,並向其中加入25 mL的冰乙酸作為溶劑,升溫至110℃,並不斷攪拌,在反應過程中,用TLC跟蹤檢測 (展開劑為乙酸乙酯和石油醚,V(乙酸乙酯):V(石油醚) = 3 : 7) ,反應進行10 h後,反應基本完全,停止反應,冷卻至室溫,在三口瓶內加入50 mL水,並置於200 mL的燒杯中,用10%的氫氧化鈉溶液調pH值至9,抽濾,得到黃色固體,乾燥;乾燥後,再溶於24 mL的乙酸乙酯中,加熱攪拌1 h後,冷卻至室溫,有大量固體析出,抽濾、乾燥,得到中間體化合物;
本步驟得到的中間體化合物的產率為80%,熔點:>300℃。用紅外光譜、核磁氫譜和核磁碳譜表徵該中間體化合物,得到的結果如下:
IR (KBr, cm–1): 3326, 3080, 1595, 1479, 1332, 1299, 760, 725. 1H NMR ( 600 MHz , DMSO ) δ: 14.33 (s, H, OH ),9.28 (s, H, NH ),8.90 (d, J = 8.40 Hz , 2H, ArH ),8.53(d, J = 7.80 Hz, 2H, ArH), 8.25 (d, J = 8.40 Hz , H, ArH), 7.80 (t, J = 7.20 Hz, 3H, ArH), 7.71 (t, J = 7.80 Hz, 2H, ArH), 7.26(d, J = 9.0 Hz, H, ArH). 13C NMR (150 MHz, DMSO) δ: 163.4, 147.7, 140.2, 128.5, 128.5, 128.4, 128.4, 127.9, 127.9, 127.9, 126.7, 126.6, 126.6, 124.5, 124.5, 124.5, 122.5, 122.5, 122.5, 118.5, 113.5.
從表徵結果知該中間體化合物的結構式為
。
二、中間體化合物的合成:
在50 mL的三口燒瓶中加入2.0 mmol中間體化合物、0.35 g雷尼鎳和30 mL的乙醇溶劑,在通入2 min氮氣後,攪拌,並用恆壓滴液漏鬥慢慢滴加7.5 mL質量百分濃度為80%的水合肼溶液,滴加完畢後,升溫至80℃回流反應5 h後,用TLC跟蹤檢測(展開劑為V(乙酸乙酯):V(石油醚) = 3 : 7),反應基本完全,冷卻至室溫後,抽濾,用乙酸乙酯洗滌五次,旋轉蒸發濾液,得到褐色固體並乾燥。再用乙酸乙酯重結晶,加熱攪拌2 h後,冷卻至室溫,有大量固體析出,抽濾、乾燥,中間體化合物。
本步驟得到的中間體化合物的產率為75%,熔點:>300℃;
用紅外光譜、核磁氫譜和核磁碳譜進行表徵,得到的結果如下:
IR (KBr, cm–1): 3377, 3318, 3207, 3067, 1616, 1547, 1499, 1249, 749, 719, 580. 1H NMR ( 600 MHz , DMSO ) δ:13.60 (s, H, OH), 12.24 (s, H, NH), 8.89 (dd, J = 8.40 Hz, 2H, ArH), 8.64 (d, J = 7.80 Hz, H, ArH),8.49 (d, J = 7.80 Hz, H, ArH), 7.78 (m, J = 7.80 Hz, 2H, ArH), 7.69 (t, J = 7.80 Hz, 2H, ArH), 7.44 (s, H, ArH), 6.84 (t, J = 7.80 Hz, H, ArH), 6.74 (d, J = 8.40Hz, H, ArH). 13C NMR (150 MHz, DMSO) δ: 170.8, 150.4, 149.5, 141.3, 134.8, 128.3, 128.1, 127.9, 127.7, 126.7, 126.2, 126.0, 124.6, 124.4, 122.7, 122.5, 122.2, 119.2, 118.0, 113.2, 110.7.
從表徵結果知該中間體化合物的結構式為
。
三、Hg2+螢光探針的合成:
依次向三口瓶中加入0.65 g(2.0 mmol)中間體化合物、0.24 g(2.0 mmol)水楊醛,再將20 mL的冰乙酸加入三口瓶中作為溶劑,常溫攪拌反應2 h,反應結束後,向反應體系中加入水淬滅反應,再置於200 mL的燒杯中,用10%氫氧化鈉溶液調pH值至10,然後進行抽濾,濾餅水洗至中性,乾燥,再用乙酸乙酯重結晶,收集固體,乾燥後,得到雙羥基菲並咪唑Hg2+螢光探針。本步驟中,雙羥基菲並咪唑Hg2+螢光探針的產率為68%。
雙羥基菲並咪唑Hg2+螢光探針的熔點為285 ~ 286℃。熔程為1℃,說明純度較高,質量百分純度大於95%。
用紅外光譜及核磁共振譜進行表徵,得到的結果如下:
IR (KBr, cm–1): 3231, 3200, 3058, 2926, 1662, 1614, 1499, 1280, 754, 723, 631. 1H NMR ( 600 MHz , DMSO ) δ:13.80 (s, H, OH ), 13.32 (s, H, NH ), 13.30 ( s, H, OH ), 9.07 ( s, H, ArH), 8.90 ( dd, J = 7.80 Hz, 2H, ArH), 8.58 ( d, J = 7.80 Hz, H, ArH), 8.51 (d, J = 7.80 Hz, H, ArH), 8.35 (s, H, ArH), 7.81 (m, J = 6.60 Hz, 2H, ArH), 7.75 (s, H, ArH), 7.68 ( d, J = 7.20 Hz, 2H, ArH), 7.29 ( d, J = 9.0 Hz, H, ArH), 7.42 (t, J = 7.20 Hz, H, ArH), 7.19 ( d , J = 9.0 Hz, H, ArH) ,7.01 (t , J = 6.60 Hz, H, ArH). 13C NMR (150 MHz, DMSO) δ: 162.8, 162.8, 161.9, 160.7, 157.2, 149.2, 140.2, 133.4, 132.7, 128.5, 128.2, 128.1, 127.8, 126.9, 126.5, 126.3, 125.9, 124.8, 124.4, 123.2, 122.7, 122.3, 122.2, 120.6, 119.9, 119.7, 118.7, 117.1,113.7.
從以上的表徵結果可知,可逆雙羥基菲並咪唑Hg2+螢光探針的結構式為:
。
將本試驗製備的可逆雙羥基菲並咪唑Hg2+螢光探針進行光譜性能測試,步驟如下:
一、儲備液的配置
將可逆雙羥基菲並咪唑螢光探針以N,N-二甲基甲醯胺(DMF)為溶劑配製成濃度為1.0×10–4 mol/L的主體儲備液,備用;
陽離子儲備液的配製:用金屬氯化鹽和硝酸鹽配製成濃度為0.10 mol/L的陽離子儲備液,備用;
陰離子儲備液的配製:用非金屬鉀鹽和鈉鹽配製成濃度為0.10 mol/L的陰離子儲備液,備用;
HEPES緩衝溶液:秤取0.60 g 的N-2-羥乙基哌嗪-N』-2-乙磺酸加入250 mL的容量瓶中,用蒸餾水定容,配成0.01 mol/L的溶液並搖勻,靜止3 h後,用氫氧化鈉溶液調pH值,配成pH值為7.4緩衝溶液。搖勻,備用。
二、光譜性能測試
向10.0 mL的容量瓶中加入濃度為1.0×10–4 mol/L的主體儲備液1.0 mL,再加入濃度為0.10 mol/L的待測金屬離子,用濃度為0.01 mol/L、pH = 7.4的HEPES緩衝溶液定容。此時,主體與Hg2+摩爾濃度比為 1 : 10。恆溫2 h後,進行螢光發射光譜的測試。
先考察可逆雙羥基菲並咪唑螢光探針對金屬離子的選擇性識別,選用的溶劑為體積比是1:1的DMF/HEPES的混合溶液(其中,HEPES緩衝溶液的濃度為0.01 mol/L,pH = 7.4)並在激發波長為315 nm,激發狹縫寬度為15 nm的情況下,測定濃度為1.0×10-5 mol/L的主體化合物的螢光強度。再向主體化合物中分別加入濃度為0.10 mol/L 的K+、Ba2+、Ca2+、Na+、Mg2+、Zn2+、Cr3+、Fe3+、Cd2+、Ni2+、Co2+、Pb2+、Cu2+、Ag+、Al3+和Hg2+金屬離子,並分別測定螢光發射光譜,結果如圖1所示。從圖1中可知,主體的螢光發射波長為495 nm,螢光強度為4 a.u.。加入不同金屬離子後,可以看出K+、Ba2+、Ca2+、Na+、Mg2+、Zn2+、Cr3+、Cd2+、Ni2+、Co2+、Pb2+、Cu2+、Ag+、Al3+ 和Fe3+與對主體的螢光強度影響不大,強度均在4 a.u.左右。而加入Hg2+時,螢光強度明顯增強為43 a.u.,增強程度為主體螢光強度的10倍。因此,從螢光發射光譜可以初步推測,主體化合物對Hg2+具有選擇識別特性。
為了進一步驗證本試驗製備的可逆雙羥基菲並咪唑螢光探針對Hg2+具有選擇性識別的特性,以體積比是1:1的DMF/HEPES的混合溶液(其中,HEPES緩衝溶液的濃度為0.01 mol/L,pH = 7.4)為溶劑,配製濃度為1.0×10-5 mol/L的主體溶液,在主體溶液中分別加入濃度為0.10 mol/L的K+、Ba2+、Ca2+、Na+、Mg2+、Zn2+、Cr3+、Cd2+、Ni2+、Co2+、Pb2+、Cu2+、Ag+、Al3+和Fe3+ 金屬離子溶液。充分混合後靜置5 min,再分別加入0.10 mol/L的Hg2+後混合均勻。此時,主體化合物、金屬離子、Hg2+三者的摩爾濃度比為1 : 10 : 10。恆溫2 h後,在激發波長為315 nm,激發狹縫寬度為15 nm的情況下,對其進行螢光發射光譜的測試。得到的螢光發射光譜強度結果如圖2所示。在其他金屬離子(K+、Ba2+、Ca2+、Na+、Mg2+、Zn2+、Cr3+、Cd2+、Ni2+、Co2+、Pb2+、Cu2+、Ag+、Al3+和Fe3+)存在的情況下,Hg2+與其他金屬離子共存時,主體化合物與汞離子識別的螢光強度並不受其他金屬離子的影響。也就是說,其他金屬離子的存在並不幹擾主體化合物對Hg2+識別。因此,圖2即可以證明主體化合物對Hg2+具有選擇識別特性。又可以說明其他金屬離子對主體化合物識別Hg2+無影響。
為了考查螢光分子-Hg2+絡合物對陰離子的選擇性識別,以體積比是1:1的DMF/HEPES的混合溶液(其中,HEPES緩衝溶液的濃度為0.01 mol/L,pH = 7.4)為溶劑,配製主體化合物溶液,在主體化合物溶液中加入0.10mol/L的Hg2+,再分別加入濃度為0.10 mol/L的AcO–、Cl–、H2PO4–、HCO3–、CO32–、HPO42–、HSO3–、S2–、NO2–、Br–、SO32–、SO42–-、BrO3–、PO43–對其進行螢光發射光譜的測試。測試結果如圖3所示,從圖3可以看出,其他陰離子的加入對該螢光探針識別Hg2+幾乎無影響,但加入S2–後螢光強度從43 a.u.減小至4 a.u.左右,說明螢光分子-Hg2+絡合物對S2–具有選擇性識別作用,完成該螢光分子探針的可逆(off-on-off)螢光信號響應。
再考察螢光分子-Hg2+絡合物對S2–具有選擇性識別特性,選用的溶劑為體積比是1:1的DMF/HEPES的混合溶液(其中,HEPES緩衝溶液的濃度為0.01 mol/L,pH = 7.4)為溶劑,配製濃度為1.0×10-5 mol/L的主體溶液,在主體溶液中分別加入濃度為0.10 mol/L的AcO-、Cl-、H2PO4-、HCO3-、CO32-、HPO42-、HSO3-、F-、NO2-、NO3-、Br-、SO32-、SO42-、BrO3-、PO43-陰離子溶液,再分別加入0.10mol/L的S2–後混合均勻。隨後對其進行螢光發射光譜的測試。測試結果如圖4所示,在其他陰離子(AcO–、Cl–、H2PO4–、HCO3–、CO32–、HPO42–、HSO3–、NO2–、Br–、SO32–、SO42–-、BrO3–和PO43–)存在的情況下,與其他陰離子共存時,主體化合物- Hg2+對S2–識別的螢光強度並不受其他陰離子的影響。也就是說,其他陰離子的存在並不幹擾S2–的識別。因此,圖4既可以證明主體化合物-Hg2+絡合物對S2–具有專一識別特性,完成該螢光分子探針的可逆 (off-on-off) 螢光信號響應,又可以說明其他陰離子對主體化合物-Hg2+識別S2–無影響。
這個結果在主體化合物的實際應用中具有重要的意義。
本試驗製備的可逆雙羥基菲並咪唑Hg2+螢光探針,通過以上試驗說明在以體積比是1:1的DMF/H2O的混合溶液(HEPES緩衝溶液的濃度為0.01 mol/L,pH = 7.4)為溶劑的體系中,該主體化合物對Hg2+有選擇性識別的作用,而對其他金屬離子無識別特性,且與其他金屬離子共存時,不幹擾該化合物識別Hg2+。該螢光分子-Hg2+絡合物還可以對S2–進行選擇性識別,即使與其他陰離子共存時也不幹擾其識別S2–。
試驗2:本試驗與試驗1不同的是試驗1的步驟三用以下的操作代替:依次向三口瓶中加入中間體化合物 0.65 g(2.0 mmol)、水楊醛 0.31 g(2.5 mmol)和20 mL的98%的濃硫酸作為溶劑,常溫攪拌反應4 h,反應結束後,向反應體系中加入水淬滅反應,並置於200 mL的燒杯中,用10%氫氧化鈉溶液調pH值至10,然後進行抽濾,濾餅水洗至中性,乾燥,再用乙酸乙酯重結晶,收集固體,乾燥後得到可逆雙羥基菲並咪唑Hg2+螢光探針。本試驗得到的雙羥基菲並咪唑Hg2+螢光探針的結構式為:
,產率為40%。
試驗3:本試驗與試驗1不同的是試驗1的步驟三用以下的操作代替:依次向三口瓶中加入中間體化合物 0.65 g(2.0 mmol)、水楊醛 0.34 g(2.8 mmol),和25 mL的苯甲酸作為溶劑,常溫攪拌反應3.5 h,反應結束後,向反應體系中加入水淬滅反應,並置於200 mL的燒杯中,用10%氫氧化鈉溶液調pH值至9,然後進行抽濾,濾餅水洗至中性,乾燥,再用乙酸乙酯重結晶,收集固體,乾燥後得到可逆雙羥基菲並咪唑Hg2+螢光探針。本試驗得到的可逆雙羥基菲並咪唑Hg2+螢光探針的結構式為:
,產率為50%。
試驗4:本試驗與試驗1不同的是試驗1的步驟三用以下的操作代替:依次向三口瓶中加入中間體化合物 0.65 g(2.0 mmol)、水楊醛 0.37 g(3.0 mmol),和15 mL的65%的濃硝酸作為溶劑,常溫攪拌反應5 h,反應結束後,向反應體系中加入水淬滅反應,並置於200 mL的燒杯中,用10%氫氧化鈉溶液調pH值至10,然後進行抽濾,濾餅水洗至中性,乾燥,再用乙酸乙酯重結晶,收集固體,乾燥後得到可逆雙羥基菲並咪唑Hg2+螢光探針。本試驗得到的可逆雙羥基菲並咪唑Hg2+螢光探針的結構式為:
,產率為45%。