一種超聲提取的川白芷多糖及製備方法和應用與流程
2023-10-10 22:33:59 7
本發明屬於生物醫藥技術領域,具體涉及一種超聲提取的川白芷多糖及製備方法和應用。
背景技術:
藥材白芷為傘形科植物白芷angelicadahurica(fisch.exhoffm)benth.ethook.f.或杭白芷angelicadahurica(fisch.exhoffm)benth.ethook.f.var.formosana(boiss)shanetyuan的乾燥根。白芷味辛,性溫,歸胃、大腸、肺經。具有解表散寒,祛風止痛,宣通鼻竅,燥溼止帶,消腫排膿的作用,用於感冒頭痛,眉稜骨痛,鼻塞流涕,鼻鼽,鼻淵,牙痛,帶下,瘡瘍腫痛。
白芷是衛生部公布藥食兩用的87種中藥材之一,也是我國常用的大宗藥材。按產地分為川、杭、祁、禹白芷四大商品。其中產於四川的白芷則稱川白芷,是著名的川產道地藥材,產量約佔全國商品白芷的70%。川白芷以四川遂寧出產的形優、質堅、粉多香濃、色白細膩、橫切片成「菊花芯形」的川白芷品質最佳。白芷富含多種化學成分,現代研究表明,白芷的脂溶性化學成分主要為香豆素類,含量為0.211%~1.221%,其水溶性成分有棕櫚酸、豆甾醇、β-谷甾醇、β-胡蘿蔔苷(β-daucosterin)等。白芷還富含多種多糖。
多糖廣泛存在於動、植物和微生物(真菌和細菌)中,常與蛋白質和多聚核苷酸相連,是生命活動中必不可少的生物大分子,在細胞吸附、細胞與細胞信息交流、免疫系統分子識別方面扮演著重要的作用。目前多糖已成為天然藥物及保健品研發中的重要組成部分。植物多糖具有多種生物活性,如抗氧化、抗腫瘤、抗病毒、降血糖血脂、抗衰老、抗炎、抗轄射、免疫調節等。
與傳統提取方法相比,採用超聲法提取多糖處理簡單安全,省時,提取效率高。但超聲波強烈的機械剪切作用,可能會致多糖分子結構的改變,使其生物活性喪失。
目前對於白芷多糖的研究尚不夠深入。僅個別學者對杭白芷多糖的提取以及抗氧化和增強動物皮膚組織免疫功能進行過分析,且尚未開展過超聲法提取的白芷多糖的結構和活性研究。
技術實現要素:
本發明的目的之一在於針對上述問題,提供一種超聲提取的川白芷多糖。
本發明的目的之二在於提供一種超聲提取的川白芷多糖的製備方法。
本發明的目的之三在於提供一種超聲提取的川白芷多糖的應用。
為實現上述目的,本發明採用的技術方案如下:
本發明所述的一種超聲提取的川白芷多糖,該川白芷多糖同時存在α構型和β構型多糖連結方式,其表觀分子量為170.07kda;所述川白芷多糖由川白芷藥材經超聲提取、醇沉除雜、去蛋白去鹽後,再經deae-瓊脂糖凝膠ff柱和瓊脂糖凝膠6ff凝膠柱層析分離而得。
本發明所述的川白芷多糖的製備方法,具體包括以下步驟:
a:脫脂脫色素:將川白芷藥材洗淨烘乾粉碎,得粉末;將所述粉末先加入石油醚脫脂後,再加入高濃度乙醇溶液脫除部分色素及低聚糖;
b:提取:將脫脂脫色素後的粉末加水超聲提取後,離心,收集上清液,濃縮,得濃縮液;
c:醇沉除雜:將所述濃縮液醇沉除去水溶性雜質後得到川白芷粗多糖;
d:去蛋白:將所述川白芷粗多糖採用酶+sevag結合法去除蛋白;
e:透析去鹽:將所述去除蛋白後的川白芷粗多糖採用透析法去除使無機鹽和小分子物質,得去鹽川白芷多糖;
f:將所述去鹽川白芷多糖進行deae-瓊脂糖凝膠ff柱層析,收集並合併第三個洗脫峰的收集液,濃縮,透析,凍幹;
g:將步驟f凍幹後的川白芷多糖進行瓊脂糖凝膠6ff凝膠柱層析,收集並合併主峰的洗脫液,濃縮,透析,凍幹,即得。
進一步地,所述步驟a中,將川白芷藥材洗淨,於40-50℃烘乾,粉碎,過40目篩,得川白芷粉末;取所述川白芷粉末加入石油醚加熱回流脫脂4h,過濾,濾渣於40-50℃烘乾後,再加入80%乙醇加熱回流提取4h,過濾,濾渣揮幹乙醇後粉碎,於40-50℃烘乾,備用;所述石油醚的用量按每克川白芷粉末計為2-4ml,所述80%乙醇的用量按每克川白芷粉末計為2ml。
進一步地,所述步驟b中加水量以每克脫脂脫色素後的粉末計為2-6ml,提取次數為1-4次,提取時間為0.5-2h。
進一步地,所述步驟d中,向濃縮液中加入無水乙醇至混合溶液乙醇終濃度為50-70%,於0-6℃靜置12-24h,離心,取離心沉澱加水溶解後,再次離心,取再次離心後的上清液,向所述上清液加入無水乙醇至混合溶液乙醇終濃度為50-70%,於0-6℃靜置12-24h,第三次離心,取第三次離心後的沉澱,得川白芷粗多糖,備用。
進一步地,所述步驟d中所用酶為木瓜蛋白酶,所用sevag試劑為氯仿:正丁醇=4:1,所述去蛋白的次數為10次。
進一步地,所述步驟e中,採用截留分子量為8000-14000da的透析袋,室溫蒸餾水透析24h後,換乾淨蒸餾水再透析8h,重複2次。
進一步地,所述步驟f中,將川白芷多糖加水溶解後,上樣於deae-瓊脂糖凝膠ff柱,先以1.5倍柱體積的蒸餾水洗脫後,再以水和2mol/l的nacl溶液混合洗脫,實現0~1.5mol/lnacl溶液的梯度洗脫,分管收集,收集液用苯酚-硫酸法檢測多糖含量,繪製苯酚-硫酸反應曲線圖,並繪製反應曲線,根據所述反應曲線合併同一洗脫峰的收集液,濃縮,透析,凍幹。
進一步地,將步驟f凍幹後的川白芷多糖加水溶解後,上樣於瓊脂糖凝膠6ff凝膠柱,洗脫液為0.05mol/lnacl溶液,分管收集,收集液用苯酚-硫酸法檢測多糖含量,繪製苯酚-硫酸反應曲線圖,採用考馬斯亮藍g250法測定蛋白含量,並繪製反應曲線,根據所述反應曲線收集主峰的洗脫液,濃縮,透析,凍幹。
本發明所述的川白芷多糖在製備具有美白作用的日化用品、食品、保健品或藥品中的用途。
本發明所用川白芷,其原植物為杭白芷[angelicadahurica(fisch.exhoffm.)benth.ethook.f.var.formosana(boiss.)shanetyuan],產地為四川省遂寧市。
與現有技術相比,本發明具備的有益效果為:
本發明提供了一種超聲提取的川白芷多糖,該川白芷多糖具有良好的美白活性,對酪氨酸酶活性具有抑制作用,且隨著濃度的增加,酪氨酸酶抑制率逐漸升高,呈現明顯的量效關係。
本發明採用超聲法提取川白芷多糖,所得川白芷多糖的分子量較大,且具有良好的美白活性,採用本發明提取方法不影響川白芷多糖的活性。經超聲法提取的川白芷粗多糖經醇沉、去蛋白去鹽後,再經兩次柱層析,得到良好的純化效果,含糖量顯著提高,澱粉幾乎除盡。
本發明建立了一條完整可行的川白芷多糖提取、分離純化、理化性質、結構和生物活性研究技術路線。
附圖說明
附圖1為本發明實施例1所得的川白芷多糖的deae-瓊脂糖凝膠ff色譜柱蒸餾水洗脫曲線圖。
附圖2為本發明實施例1所得的川白芷多糖的deae-瓊脂糖凝膠ff色譜柱nacl梯度洗脫曲線圖。
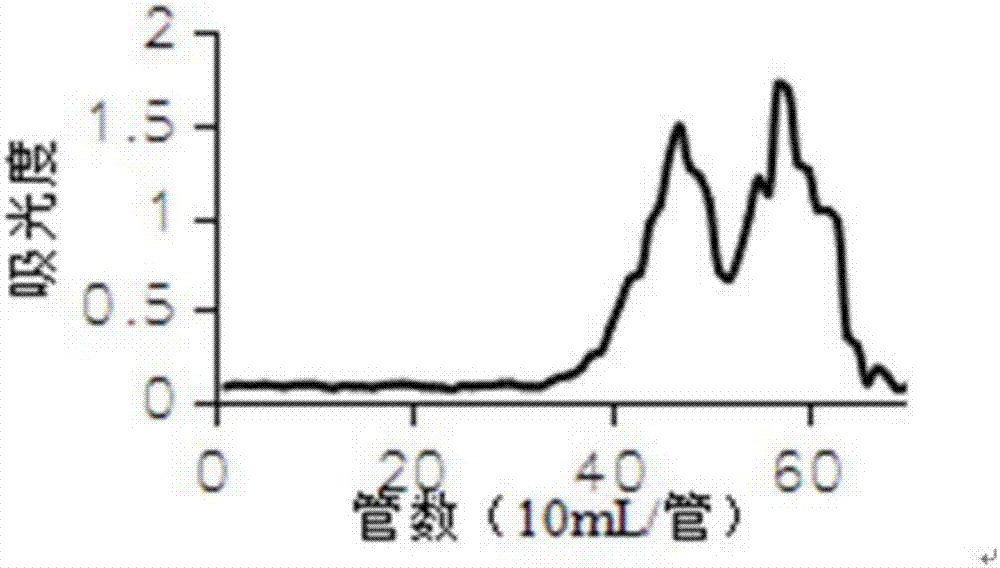
附圖3為本發明實施例1所得的川白芷多糖的瓊脂糖凝膠6ff色譜柱洗脫曲線圖。
附圖4為本發明實施例1所得的川白芷多糖的剛果紅反應曲線圖。
附圖5為本發明實施例1所得的川白芷多糖的傅立葉變換紅外光譜圖。
附圖6為本發明實施例1所得的川白芷多糖的1hnmr圖譜,
附圖7為本發明實施例1所得的川白芷多糖的為13cnmr圖譜。
附圖8為本發明實施例1所得的川白芷多糖的酪氨酸酶抑制活性圖。
附圖9為本發明二次過柱的川白芷多糖的酪氨酸酶抑制活性圖。
具體實施方式
下面結合附圖說明和實施例對本發明作進一步說明,本發明的方式包括但不僅限於以下實施例。
實施例1
川白芷多糖的製備
本實施例中超聲提取所用設備為sb25-12dtd超聲清洗機(寧波新芝生物科技股份有限公司)。
川白芷多糖的製備方法,包括以下步驟:
a:脫脂脫色
取川白芷根洗淨,45℃烘乾,粉碎過40目篩。稱取白芷粉樣,加入石油醚回流脫脂4h,45℃烘乾後,再加入80%乙醇80℃回流提取4h,以除去部分色素及低聚糖,揮幹乙醇後粉碎,45℃烘乾備用。石油醚的用量按每克川白芷粉末計為3ml,80%乙醇的用量按每克川白芷粉末計為2ml。
b:提取
稱取脫脂脫色素後的粉末100g,加入3倍量蒸餾水超聲提取2次,每次1h,4000r/min離心5min收集上清液,合併提取液並濃縮,得濃縮液。
c:醇沉除雜
向所述濃縮液中緩慢加入無水乙醇至混合溶液乙醇終濃度為60%,置於4℃靜置12h,離心得川白芷多糖沉澱,棄上清液。川白芷多糖沉澱用水復溶,4000r/min離心10min除雜質,得上清液加乙醇濃度至60%,4℃靜置12h後,第三次離心,取第三次離心後的沉澱,備用;
d:去蛋白
按照lg川白芷多糖沉澱加入200ml水的比例溶解樣品,在50℃水浴加熱並間歇性攪拌至其完全溶解,4000r/min離心5min除去雜質,川白芷多糖溶液ph調節到6.5。木瓜蛋白酶用ph6.5的pbs配成50mg/ml的酶溶液,按照lg川白芷多糖沉澱加20mg木瓜蛋白酶的比例加到上述的糖溶液中,然後在50℃水浴加熱3h,再在100℃下滅酶15min,冷卻至室溫,4000r/min離心10min除去變性蛋白和酶。
在經過木瓜蛋白酶解後的混合溶液中加入等體積的sevag試劑(氯仿:正丁醇=4:1),劇烈振搖15min,5000r/min離心10min,離心後液體三分層,小心吸取上層清液並重複去蛋白10次後40℃旋轉蒸發真空濃縮得到去蛋白川粗多糖。
e:透析去鹽
8000-14000da透析袋處理液為0.01mol/lnahco3和lmmol/ledta的水溶液。將長度為25cm左右透析袋放入處理液加熱煮沸半個h。蒸餾水清洗透析袋,置於50%乙醇中,4℃保存備用。將去蛋白川粗多糖溶液適量裝於透析袋中,透析袋兩頭紮好後留出1/3的空間防止其中親水性物質過度吸水使袋破裂。室溫下用蒸餾水透析24h後,換乾淨蒸餾水再透析8h,重複2次。透析後濃縮、凍幹得到川白芷粗多糖。
f:deae-瓊脂糖凝膠ff柱層析
首先,將500ml的deae-瓊脂糖凝膠ff裝入2.5×100cm的中壓層析柱中。柱子裝好後用3倍柱體積的去蒸餾水,1ml/min流速進行平衡。
其次,將川白芷粗多糖配製成50mg/ml的水溶液樣品,所有備用洗脫液要超聲脫氣,上樣量5ml,流速lml/min,先以1.5倍柱體積的去蒸餾水洗脫,再以水和2mol/l的nacl混合洗脫,實現0~1.5mol/lnacl溶液的梯度洗脫,每10ml收集一管。每隔一管取樣100μl用苯酚-硫酸法(100μl樣品溶液+200μl5%苯酚溶液+1ml濃硫酸)檢測多糖含量,繪製苯酚-硫酸反應曲線圖,並繪製反應曲線。收集並合併第3個洗脫峰的收集液,濃縮,透析,凍幹。川白芷多糖deae-瓊脂糖凝膠ff色譜柱洗脫曲線如附圖1和附圖2所示。
g:瓊脂糖凝膠6ff凝膠柱層析
將步驟f凍幹後的川白芷多糖過瓊脂糖凝膠6ff凝膠柱進行過濾層析,配製成30mg/ml的多糖水溶液,上樣量10ml,洗脫液為0.05mol/lnacl溶液,流速為0.5ml/min,每管收集5ml。每管取樣100μl用苯酚硫酸法檢測多糖,繪製苯酚-硫酸反應曲線圖,採用考馬斯亮藍g250法測定蛋白含量,並繪製反應曲線。收集主峰的洗脫液,濃縮,透析,凍幹,即得。川白芷多糖初步分離組分的瓊脂糖凝膠7ff色譜柱洗脫曲線如附圖3所示。
實施例2
川白芷多糖的製備
本實施例中超聲提取所用設備為sb25-12dtd超聲清洗機(寧波新芝生物科技股份有限公司)。
川白芷多糖的製備方法,包括以下步驟:
步驟a:與實施例1的步驟a相比,烘乾溫度均為50℃,石油醚的用量按每克川白芷粉末計為2ml,其餘條件均相同。
步驟b:稱取脫脂脫色素後的粉末100g,加入2倍量蒸餾水超聲提取2h,4000r/min離心5min收集上清液,合併提取液並濃縮,得濃縮液。步驟c至步驟g同實施例1,即得。
實施例3
川白芷多糖的製備
本實施例中超聲提取所用設備為sb25-12dtd超聲清洗機(寧波新芝生物科技股份有限公司)。
川白芷多糖的製備方法,包括以下步驟:
步驟a:與實施例1的步驟a相比,烘乾溫度均為40℃,石油醚的用量按每克川白芷粉末計為4ml,其餘條件均相同。
步驟b:稱取脫脂脫色素後的粉末100g,加入6倍量蒸餾水超聲提取4次,每次0.5h,4000r/min離心5min收集上清液,合併提取液並濃縮,得濃縮液。步驟c至步驟g同實施例1,即得。
實施例4
對實施例1得到的川白芷多糖進行總糖檢測,檢測方法如下:
1mg/ml葡萄糖母液的配置:葡萄糖在烘箱中100℃烘乾至恆重,稱0.1033g葡萄糖於100ml容量瓶中,加蒸餾水溶解定容,備用。
葡萄糖標準曲線繪製:分別取適量1mg/ml葡萄糖母液,稀釋至葡萄糖濃度為0.1、0.2、0.4、0.6、0.8、1mg/ml,吸取100μl葡萄糖溶液,然後加入5%苯酚200μl及1ml濃硫酸,搖勻,室溫放置20min後於490nm下測吸光度。以加入葡萄糖濃度為橫坐標,490nm下吸光度為縱坐標繪製葡萄糖標準曲線。
取實施例1得到的川白芷多糖,配製1mg/ml的不同樣品溶液,取100μl檢測,做三組平行試驗取平均值。
以葡萄糖為標準品繪製標準曲線,苯酚-硫酸法檢測總糖的含量,回歸方程為y=2.6803x+0.1572,r2=0.994。根據樣品測定的吸光度值對照標準曲線回歸方程計算白芷多糖總糖含量,得到實施例1所得的川白芷多糖中總糖的含量為46.01%。
實施例5
對實施例1得到的川白芷多糖進行蛋白質分析,實驗方法如下:
考馬斯亮藍g250是一種蛋白質染料,與蛋白質結合使之染色在595nm處有最大吸收,常用來定性和定量測定蛋白質,該方法操作簡單、反應迅速適合大量樣品的測定,在0-100mg/l蛋白質濃度範圍內,吸光值與蛋白質含量呈良好的線性關係。
1mg/ml樣品的水溶液,可見光-紫外光分光光度計進行400-200nm紫外掃描,以蒸餾水為空白對照。
蛋白質標準曲線繪製:稱100mg考馬斯亮藍g250溶於50ml95%乙醇,加入100ml85%磷酸,蒸餾水定容至1l,濾紙過濾備用。配製0.1mg/ml牛血清白蛋白標準溶液,分別取0ml、0.2ml、0.4ml、0.6ml、0.8ml、1.0ml,用蒸餾水補充至1.0ml,分別加入考馬斯亮藍g250溶液各5ml,混勻。放置2min後在595nm下測吸光度。
配製成lmg/ml的不同樣品溶液,取lml檢測,做三組平行試驗取平均值。
以牛血清白蛋白為標準品繪製標準曲線,線性回歸方程為:y=0.0025x+0.006,r2=0.9907。根據樣品測定的吸光度值對照標準曲線回歸方程計算白芷多糖蛋白質含量,得到實施例1所得的川白芷多糖中蛋白質含量為0.56%。由於測量存在一定誤差,該值並不能代表樣品蛋白質含量,只能說明樣品中不含蛋白質或可能含有結合蛋白。
實施例6
對實施例1得到的川白芷多糖進行糖醛酸定性分析,實驗方法如下:
咔唑試液的製備:精密稱取0.125g咔唑,加無水乙醇溶解定容至100ml的量瓶中,備用。
配製0.4mg/ml葡萄糖醛酸標準溶液,1mg/ml的樣品溶液。
取50μl樣品溶液,加入50μl咔唑試液,冰浴加入300μl濃硫酸,觀察顏色變化。
結果表明:實施例1得到的川白芷多糖醛糖酸檢測與400μg/mld-葡萄糖醛酸顯色一致,均呈藍綠色陽性顯色,說明其可能含有醛糖酸,是酸性雜多糖。
實施例7
對實施例1最終得到的川白芷多糖進行碘-碘化鉀反應檢測,方法如下:
碘試劑為含0.02%碘的0.2%ki溶液,川白芷多糖及水提粗多糖的樣品濃度均為lmg/ml。分別取50μl各樣品溶液,加入200μl碘試劑,觀察顏色變化後用酶標儀(thermoscientificmultiskango)進行200—800nm掃描。
結果顯示,川白芷多糖與碘試劑反應呈陰性,說明通過兩次過柱分離純化後川白芷多糖中澱粉已幾乎除盡。
將川白芷多糖樣品溶液與碘試劑(含0.02%碘的0.2%ki溶液)混勻,測定300-700nm範圍內的吸收光譜,最大吸收峰在351nm附近,而565nm附近並沒有最大吸收,說明川白芷多糖可能存在較長的側鏈和較多的分支。
實施例8
對實施例1得到的川白芷多糖進行剛果紅試驗,具體實施方法如下:
配置1mg/ml川白芷多糖溶液,80μmol/l剛果紅溶液,1mol/l的naoh溶液。
吸取0.5ml川白芷多糖溶液,加入80μmol/l剛果紅溶液0.5ml,混勻。然後加入適量1mol/l的naoh溶液,使反應溶液中naoh的終濃度分別為0.0mol/l、0.1mol/l、0.2mol/l、0.3mol/l和0.4mol/l混勻室溫靜置後,200-800nm區間進行光譜掃描,測量不同濃度下溶液的最大吸收波長。另取空白對照,即不加川白芷多糖樣品,以相同方法測量不同濃度下溶液的最大吸收波長。
比較樣品溶液與空白對照溶液的最大吸收波長變化情況。
結果如附圖4所示,隨著naoh濃度從低到高,川白芷多糖最大吸收波長變化情況與空白對照溶液變化趨勢基本相同,說明其可能不具有三股螺旋結構。
實施例9
對實施例1得到的川白芷多糖進行多糖分子量分析,具體實驗方法如下:
採用高效液相色譜-蒸發光散射檢測法(hplc-elsd)對川白芷多糖分子量進行測定。hplc色譜條件如下:色譜柱:tsk-gelg5000pwxl凝膠柱(7.8mm×30cm);流動相:水;流速:0.5ml/min;進樣量:20μl;檢測器:alltech2000型elsd檢測器,漂移管溫度:115℃,氣體流量:3.2(slpm)。
標準曲線的製備:取平均分子量分別為t10、t40、t70、、t500、t2000道爾頓的標準葡聚糖對照品,以水溶解,配製為2mg/ml的標準溶液。照上述hplc色譜條件檢測,以標準葡聚糖的保留時間和分子量分別取自然對數繪製。
用高純水將多糖樣品製成2mg/ml的溶液,在相同色譜條件下進樣分析,記錄保留時間,帶入回歸方程算得平均分子量。
標準多糖參照品:blue2000(mw:2000kda),t-700(mw:700kda),t-500(mw:500kda),t-70(mw:70kda)t-40(mw:40kda),t-10(mw:10kda)經hplc‐elsd分析後。以保留時間對分子量對數作圖,得到回歸方程y=-1.99x+21.104,r2=0.9991。由回歸方程可以計算得到川白芷多糖的表觀分子質量。
結果表明,川白芷多糖的表觀分子質量為170.07kda。
實施例10
對實施例1得到的川白芷多糖進行紅外光譜和核磁共振分析
將乾燥的川白芷多糖樣品3.0mg與乾燥的kbr在瑪瑙研缽中研磨均勻後壓片,在4000-400cm-1,範圍內進行掃描。取20mg川白芷多糖溶於0.5ml重水中,核磁共振儀進行600mhznmr分析。
如附圖5紅外光譜圖顯示,1022.20cm-1、1085.85cm-1、1095.49cm-1三處有吸收峰,表明川白芷多糖的糖環結構為呋喃糖型。在846.692cm-1處有吸收峰說明其存在α構型的多糖連結方式。在891±7cm-1的吸收峰說明存在β構型多糖連結方式。
在1647.095cm-1附近的吸收峰表明在川白芷多糖各組分中存在乙醯氨基(-nhcoch3)的c=o鍵伸縮振動,說明川白芷多糖可能是一種氨基多糖。在1733.886cm-1和1261.359cm-1為醯基或o-乙醯基(o-ac)的特徵吸收峰。
綜上,川白芷多糖為同時存在α構型和β構型多糖連結方式,具有醯基或o-乙醯基(o-ac)的一種呋喃糖型氨基多糖片段。
川白芷多糖的1hnmr如附圖6所示,13cnmr如圖7所示。β型異頭質子區域化學位移為δ4.4-5.0ppm,而α型異頭質子區域化學位移為δ5.0-5.5ppm。在1hnmr信號指紋區δ4.4-5.5ppm,有信號δ4.43、δ4.91、δ5.04、δ5.06、δ5.10、δ5.19和δ5.34ppm,可見川白芷多糖有至少7種單糖殘基。h信號δ4.43、δ4.91表示多糖為存在β構型連結糖殘基,而h信號δ5.04-5.34表明川白芷多糖也存在α構型連結糖殘基。在13cnmr中,在δ90-112ppm區域,有δ107.47、103.73、103.43、102.66、99.80ppm,其中δ107.47、103.73、103.43ppm表示存在以β構型連結的糖殘基,而其他共振信號表示川白芷多糖存在α構型連結糖殘基,這與1hnmr分析結果一致。在1hnmr,δ4.68ppm的共振峰為doh吸收峰,而在δ3.5-4.4ppm之間的信號主要是由於糖殘基上c2-c6上的h信號位移峰,在δ3.3-3.5ppm之間,有h信號,表示存在meo,在δ2.1ppm附件的共振峰測表示存在n-ac。在13cnmr中,在δ170-180ppm之間有共振峰,表示存在糖醛酸,δ70-75ppm的共振區為未發生取代多糖部位(c2、c3、c4)的碳信號,而在δ76-85ppm的c信號表示c2、c3、c4上發生取代,而在δ67ppm附近有c信號,表示c6位也發生了取代,在δ60ppm附近的c信號表示存在meo基團,在δ52.90ppm為n取代上c的信號。
綜上,川白芷多糖可能為多種糖殘基組成並且同時存在α構型和β構型連結糖殘基的酸性雜多糖,還有meo、n-ac(n-乙醯基)、糖醛酸等基團,在c2、c3、c4、c6均可能存在取代。
實施例11
對實施例1得到的川白芷多糖進行美白活性分析,具體實驗方法如下:
採用酪氨酸酶多巴速率氧化法,以酪氨酸酶抑制率為指標進行評價,具體操作如下。
試劑配製
(1)pbs緩衝液配製
0.2mol/l的nah2po4:稱取nah2po4·h2o3.12g加蒸餾水定容至100ml溶解。
0.2mol/l的na2hpo4:稱取na2hpo4·2h2o3.56g加蒸餾水定容至100ml溶解。
0.1mol/l的pbs,49ml0.2mol/l的na2hpo4,51ml0.2mol/l的na2hpo4,加100ml蒸餾水稀釋。
(2)多巴配製
取0.075g左旋多巴,用pbs緩衝液定容到100ml容量瓶。
(3)酪氨酸酶配製
取0.5mg的酪氨酸酶,用pbs緩衝液溶解,定容值2ml。酶活力單位在250u/ml左右。
(4)樣品處理
將待測樣品配置成1mg/ml、0.5mg/ml、0.25mg/ml,0.125mg/ml,0.0625mg/ml,0.03125mg/ml、0.01625mg/ml,相應濃度曲酸做陽性對照,反應體系如下表。
酪氨酸酶抑制反應體系表
按照順序加入多巴、pbs、多糖/曲酸,加入酶液後迅速放入37℃的酶標儀孵育5min測值,每個樣品做三個重複。
a:處理組樣品在475nm處的吸光度;
a0:空白組樣品在475nm處的吸光度;
b:對照組樣品在475nm處的吸光度;
b0:總空白組樣品在475nm處的吸光度。
如附圖8所示,結果表明,本發明川白芷多糖對酪氨酸酶活性具有明顯的抑制作用,美白作用確切;且隨著濃度的增加,酪氨酸酶抑制率逐漸升高,呈現明顯的量效關係。
實施例12
將實施例1得到的川白芷多糖再次過瓊脂糖凝膠6ff凝膠柱,實驗操作及條件同實施例1中的步驟g,得到二次過柱後的川白芷多糖。
將此二次過柱後的川白芷多糖進行美白活性分析,具體實驗方法同實施例11。
結果如附圖9所示,二次過柱後的川白芷多糖的美白作用明顯增強,表現出幾乎與對照曲酸相當的美白作用;且隨著濃度的增加,酪氨酸酶抑制率逐漸升高,呈現明顯的量效關係。
上述實施例僅為本發明的優選實施方式之一,不應當用於限制本發明的保護範圍,但凡在本發明的主體設計思想和精神上作出的毫無實質意義的改動或潤色,其所解決的技術問題仍然與本發明一致的,均應當包含在本發明的保護範圍之內。