抗氧化型多甲基側鏈二氟單體及其製備方法與流程
2023-05-16 01:40:37 2
本發明屬於材料技術領域,具體涉及一種抗氧化型多甲基側鏈二氟單體及其製備方法。
背景技術:
陰離子交換膜經過長期發展,現已廣泛應用於滲析、膜電解、電化學傳感器、燃料電池等領域。其中燃料電池用陰離子交換膜作為一種新型的能源功能材料,因其成本低、甲醇滲透率低等優點,具有廣泛的產業化應用前景。
陰離子交換膜通過功能性單體化合物的聚合製備,按結構可分為主鏈型聚合物和側鏈型聚合物。通過調控單體化合物或聚合物的化學結構的手段,可以改善陰離子交換膜的性能。
研究表明,與主鏈型聚合物相比,側鏈型聚合物的官能團遠離主鏈,能有效形成相分離結構,從而增大離子傳輸,提高電導率。例如,發明人所在的課題組前期授權的專利CN 104326893 A中公布的單體化合物,由於擁有較長的側鏈結構,用其製備的陰離子交換膜的電導率表現優異。通常,增多單體的可離子化基團可提高電導率,但是隨著離子化程度的提高,膜的力學性能下降,溶脹率增加,穩定性會降低。發明人所在的課題組前期授權的專利CN 104262211 A中公布了一種具有偶醯基團的單體化合物,發現利用可交聯偶醯基團製備出的交聯膜,其穩定性明顯優於未交聯膜。
技術實現要素:
本發明所要解決的技術問題在於針對陰離子膜電導率低、穩定性不足的問題,提供一種含有偶醯官能團和酚羥基的抗氧化型多甲基側鏈二氟單體,以及該單體的合成方法。
解決上述技術問題所採用的技術方案是該抗氧化型多甲基側鏈二氟單體的結構式如下所示:
其中,X代表CH3,n為1~3的整數。
本發明抗氧化型多甲基側鏈二氟單體優選下述式I~VIII化合物中的任意一種:
上述抗氧化型多甲基側鏈二氟單體的合成路線為:
具體合成步驟如下:
1、製備化合物a
在氮氣保護下,將4-甲氧基氯苄、鎂粉混合攪拌,製備成格氏試劑;將格氏試劑與2,6-二氟苯腈在室溫下攪拌反應3~4小時,停止反應,用鹽酸水解,分離純化產物,製備成化合物a。
2、製備化合物b
將化合物a與溴化銅在70~90℃下反應7~9小時,反應結束後,分離純化產物,製備成化合物b。
3、製備化合物c
將化合物b在氫溴酸和醋酸的混合體系中回流反應11~14小時,反應結束後,分離純化產物,製備成化合物c。
4、製備化合物d
將化合物c與N-溴代丁二醯亞胺(NBS)在25~30℃下反應5~7小時,反應結束後,分離純化產物,製備成化合物d。
5、製備抗氧化型多甲基側鏈二氟單體
在氮氣保護下,將化合物d、化合物e、碳酸鈉、四(三苯基)磷鈀、四丁基溴化銨按摩爾比為1:2~2.4:3~6:0.03~0.06:0.05~0.15,回流反應10~12小時,反應結束後,分離純化產物,製備成抗氧化型多甲基側鏈二氟單體。
上述步驟1中,優選4-甲氧基氯苄、鎂粉、2,6-二氟苯腈的摩爾比為1:1.2:0.8。
上述步驟2中,優選化合物a與溴化銅的摩爾比為1:1.5。
上述步驟4中,優選化合物c與N-溴代丁二醯亞胺的摩爾比為1:2。
上述步驟5中,優選化合物d與化合物e、碳酸鈉、四(三苯基)磷鈀、四丁基溴化銨的摩爾比為1:2.2:4:0.05:0.1。
本發明的有益效果如下:
1、本發明單體含有可聚合二氟基團和可功能化的甲基,其經過縮聚反應和功能團轉化後可以得到季銨化的聚合物,該功能化聚合物可作為燃料電池聚合物電解質膜材料
2、本發明單體擁有較長的側鏈結構及較多的離子化官能團,保證了它能夠擁有較高的電導率。
3、本發明單體含有偶醯基團及活性較高的酚羥基,利用偶醯基團與氨基的交聯和酚羥基能夠捕捉含氧自由基而終止氧化的作用,可用於製備擁有較高電導率且穩定性良好的抗氧化型陰離子膜。
附圖說明
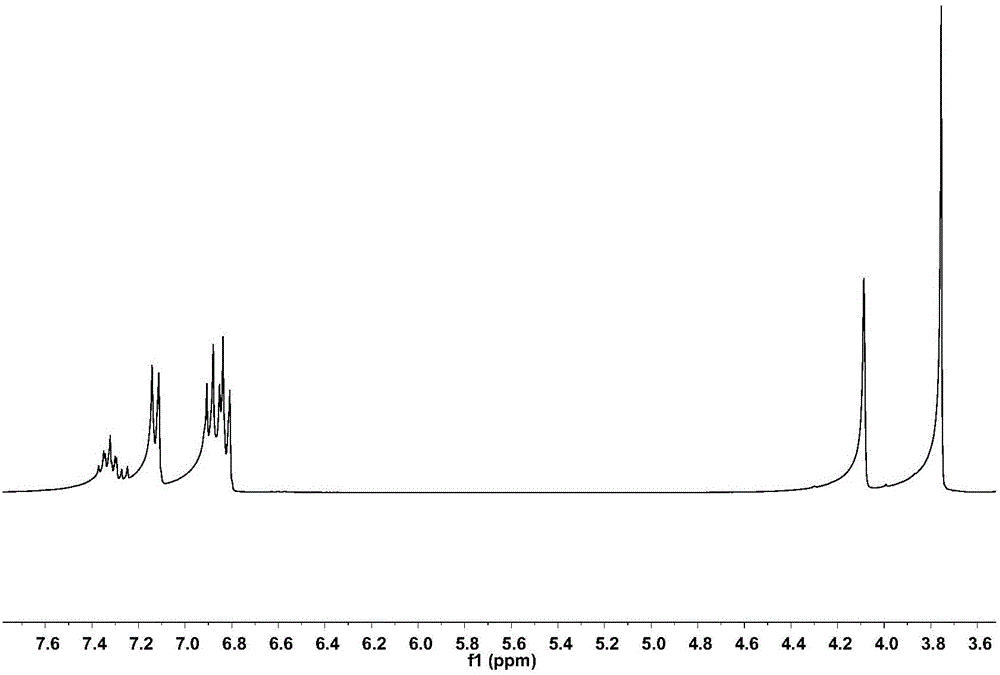
圖1是化合物a的1H核磁共振譜圖。
圖2是實施例1製備的四甲基四苯環側鏈型二氟單體的1H核磁共振譜圖。
圖3是實施例1製備的四甲基四苯環側鏈型二氟單體的質譜圖。
圖4是實施例1製備的四甲基四苯環側鏈型二氟單體的紅外光譜圖。
具體實施方式
下面結合附圖和實施例對本發明進一步詳細說明,但本發明的保護範圍不僅限於這些實施例。
實施例1
製備結構式如下的四甲基四苯環側鏈型二氟單體(式VI化合物)
1、製備化合物a
在氮氣保護下,向250mL三口燒瓶中加入6.44g(268.18mmol)鎂粉和15mL乾燥的四氫呋喃、15mL乾燥的乙醚,並加入一粒碘引發,開啟磁力攪拌,升溫至30℃,緩慢滴加35g(223.49mmol)4-甲氧基氯苄與175mL四氫呋喃、175mL乙醚的混合溶液,觀察反應現象,待格氏引發後移至低溫反應浴中於-8℃繼續滴加剩餘溶液。滴加完成後繼續反應3小時,製備成格氏試劑。然後加入23.80g(178.79mmol)2,6-二氟苯腈與80mL乾燥的四氫呋喃,室溫反應3.5小時。反應結束後,加入400mL 1mol/L的鹽酸水溶液,回流反應10小時,待反應完成後,將反應液水洗至中性,濃縮有機相,得到化合物a,其收率為21%,結構表徵數據為:
1H NMR(CHCl3-d6為溶劑,TMS為內標,400MHz,ppm):7.30(m,1H),7.18(s,2H),6.88(t,2H),6.81(d,2H),4.05(d,2H),3.76(s,3H)(具體見圖1)。
2、製備化合物b
向500mL單口燒瓶中加入12.78g(57.20mmol)溴化銅、10g(38.13mmol)化合物a和120mL乙酸乙酯、120mL二甲基亞碸,開啟磁力攪拌,升溫至80℃反應8小時,反應完成後,將反應液減壓濃縮,用無水乙醇重結晶,得到化合物b,其收率為78%。
3、製備化合物c
向500mL單口燒瓶中加入10g(35.94mmol)化合物b、120mL氫溴酸和120mL醋酸,開啟磁力攪拌,回流反應12小時。反應完成後,將體系冷卻至室溫,倒入250mL蒸餾水中攪拌,將析出的固體過濾收集,加入100mL二氯甲烷攪拌10小時,得到化合物c,其收率為71%。
4、製備化合物d
向500mL單口燒瓶中加入13.58g(76.27mmol)N-溴代丁二醯亞胺、10g(38.14mmol)化合物c和300mL四氫呋喃,開啟磁力攪拌,在28℃下反應6小時。反應完成後,將反應液減壓濃縮,濃縮後粗品進行乾燥,加入蒸餾水60℃加熱攪拌6小時,攪拌完成後過濾出固體,用乙酸乙酯重結晶,得到化合物d,其收率為55%。
5、製備四甲基四苯環側鏈型二氟單體
在氮氣保護下,向250mL三口燒瓶中加入5g(11.90mmol)化合物d、5.05g(47.6mmol)碳酸鈉,3.93g(26.19mmol)3,5-二甲基苯硼酸、0.69g(0.60mmol)四(三苯基)磷鈀、0.38g(1.19mmol)四丁基溴化銨和100mL甲苯、100mL蒸餾水、25mL乙二醇二甲醚,開啟磁力攪拌,加熱回流反應10小時。反應完成後,將反應液過濾,用二氯甲烷萃取,有機相經減壓濃縮、柱層析純化(展開劑為石油醚與二氯甲烷體積比60:1的混合液),柱層析後用乙醇重結晶,得到式IV化合物,其收率為31%,結構確認結果見圖2~4,具體數據如下:
1H NMR(DMSO-d6為溶劑,TMS為內標,400MHz,ppm):9.62(s,1H),7.85(s,2H),7.79(m,1H),7.29(t,2H),7.18(s,4H),7.02(s,2H),2.33(s,12H)(具體見圖2)。
EI-MS m/z(rel.int.):329.18(100),141.05(5.61)(具體見圖3)。
IR(KRr壓片,cm-1):3456,3026,2921,1668,1598,1465,1465,1396,1346,1240,1182,1001,854(具體見圖4)。
實施例2
製備結構式如下的四甲基四苯環側鏈型二氟單體(式IV化合物)
在實施例1的步驟5中,所用的3,5-二甲基苯硼酸用等摩爾2,4-二甲基苯硼酸替換,其他步驟與實施例1相同,得到式IV化合物,其收率為33.1%。
實施例3
製備結構式如下的二甲基四苯環側鏈型二氟單體(式III化合物)
在實施例1的步驟5中,所用的3,5-二甲基苯硼酸用等摩爾4-甲基苯硼酸替換,其他步驟與實施例1相同,得到式III化合物,其收率為38.1%。
實施例4
製備結構式如下的二甲基四苯環側鏈型二氟單體(式I化合物)
在實施例1的步驟5中,所用的3,5-二甲基苯硼酸用等摩爾的2-甲基苯硼酸替換,其他步驟與實施例1相同,得到式I化合物,其收率為31.3%。
實施例5
製備結構式如下的二甲基四苯環側鏈型二氟單體(式II化合物)
在實施例1的步驟5中,所用的3,5-二甲基苯硼酸用等摩爾的3-甲基苯硼酸替換,其他步驟與實施例1相同,製得到式II化合物,其收率為30.8%。
實施例6
製備結構式如下的四甲基四苯環側鏈型二氟單體(式V化合物)
在實施例1的步驟5中,所用的3,5-二甲基苯硼酸用等摩爾的3,4-甲基苯硼酸替換,其他步驟與實施例1相同,得到式V化合物,其收率為31.8%。