一種網絡結構成炭劑及其製備方法和應用與流程
2023-05-01 02:04:06 2
本發明屬於高分子材料技術領域,涉及一種網絡結構成炭劑,具體涉及一種含哌嗪和三聚氰胺的網絡結構成炭劑及其製備方法和應用,尤其是在製備阻燃高分子材料中的應用。
背景技術:
隨著高分子材料工業的發展,合成材料已廣泛應用於國民經濟和人民生活的各個領域。但是,由於高分子材料的易燃性和可燃性而引起的火災事故也給人類的生命財產安全帶來了巨大的威脅。在樹脂中添加阻燃劑,是提高高分子材料阻燃性能最簡單、經濟和富有成效的方法,其中阻燃劑是決定材料阻燃性能優劣的關鍵。傳統的滷系阻燃劑燃燒時釋放出滷化氫、二噁英等有毒和腐蝕性氣體,產生較大的煙霧,已經逐步被限制使用。
隨著人們環保意識的提高,阻燃劑及阻燃材料的無滷化成為科學研究和工業應用共同追求的目標。在無滷阻燃劑中膨脹型阻燃劑(IFR)因其低毒、低煙、低腐蝕、抗融滴等特點被認為是具有良好發展前景的綠色阻燃劑之一。IFR一般由酸源、碳源、氣源三部分組成,在燃燒過程中IFR可以促使材料表面形成膨脹炭層,起到隔熱、隔氧、抑煙的作用,從而使火焰熄滅。目前可用於IFR碳源的成炭劑主要存在以下問題:季戊四醇(PER)的熱穩定性較差,吸水性較大,而且在加工和使用過程中易遷出、易反應,最終導致阻燃聚合物易吸潮,進而在日常使用過程中影響產品的電性能、耐候性以及耐久性,最終影響材料使用性能。澱粉、多糖類生物基成炭劑阻燃效率低,且不耐溫。這些問題阻礙了IFR在很多領域的工業化應用。因此開發新型成炭劑是IFR發展的一個重要方向。
近年來針對傳統成炭劑的缺點,各種新型成炭劑相繼被研究報導出來。如:公開號為:CN104693442A和CN103756015等公開了利用含磷二胺或者二羥基化合物與三聚氯氰進行反應製備了一種超支化成炭劑,雖然製備成炭劑的成炭效率高,產物的熱穩定性好,但是原料中使用了三聚氯氰,需要進行階段控溫,並且需要加入縛酸劑來保證反應的順利進行,合成工藝相對繁雜,而且氯容易殘留。另有公開號為:CN103992481A公開了「一種超支化聚磷腈阻燃成炭劑及其製備方法」,雖然製得的成炭劑阻燃效果好,加工性能良好,但是,該成炭劑的製備需要20h左右,並且原料六氯環三磷腈易升華,其蒸汽對眼睛和呼吸道有刺激作用,且對水資源有害。
本專利以哌嗪和醛類化合物作為單體,以三聚氰胺為交聯劑,在溶劑中進行反應,最後以單官能矽烷偶聯劑來封端,製備了具有網絡結構的成炭劑。該成炭劑不但具有良好的熱穩定性和耐水性,而且具有功能性表面,與聚合物相容性好,此外製備方法簡單、環保,產率高,易於工業化生產,所用原材料價格便宜。可作為IFR的成炭劑用於高分子材料的阻燃,與現有的成炭劑比有明顯的優勢。
技術實現要素:
本發明的一個目的是提供一種新型網絡結構成炭劑。該成炭劑不僅成炭性能優異,而且製備方法簡單,產率高,工藝過程環保,易於工業化生產。
本發明所述的成炭劑分子結構如式(1):
式中1<n<10000;R的結構為下列之一:R1為含單個羥基或氨基的矽烷偶聯劑基團。
本發明的另一目的是提供一種製備該網絡結構成炭劑的方法。該方法具體步驟如下:
步驟(1)、將醛基化合物加入到有機溶劑中,調節溶液PH至8~9;其中醛基化合物的濃度為0.6L/mol;
所述的用於調節溶液PH的溶液為有機鹼或無機鹼中的一種;
所述的有機溶劑為N-N二甲基甲醯胺、N-N二甲基乙醯胺、四氫呋喃、二氧六環、乙腈、二甲亞碸中的一種;
所述的含醛基化合物為下列之一:甲醛溶液、固體甲醛、乙醛、1-丁醛、苯甲醛、糠醛;
步驟(2)、將哌嗪溶解於蒸餾水中得到哌嗪水溶液,滴加於步驟(1)的溶液中,滴加時間為0.5~1h,反應溫度控制在0~60℃,反應時間2~10h,得到含有A結構的透明溶液;
所述的無水哌嗪的分子式為C4H10N2;
所述的哌嗪與醛類化合物的投料物質的量比為1:2~3;
所述的哌嗪溶液的濃度為40~50%;
所述A的結構為:其中R的結構為下列之一:
步驟(3)、向步驟(2)所得的溶液中,加入三聚氰胺,溫度控制在0~60℃,反應時間1~5h,得到含有B結構的溶液;
所述的三聚氰胺的用量為哌嗪物質的量的1~10%;
所述的B結構如下:
其中R的結構為下列之一:
步驟(4)、向步驟(3)中的溶液中,再次滴加哌嗪的水溶液,溫度控制在60~90℃,反應時間2~10h,得到白色固液混合物;
所述的哌嗪投料量與步驟(2)中的哌嗪等量;
步驟(5)、向步驟(4)中的固液混合物中,加入單官能矽烷偶聯劑反應1~2h,反應結束後,經過抽濾、水洗、乾燥,得到目標產物網絡結構成炭劑。
所述的偶聯劑的用量為0.5~1%(以總溶液質量計);
所述的矽烷偶聯劑為含單個羥基或氨基的矽烷偶聯劑,具體是下列偶聯劑之一:氨丙基三乙氧基矽烷、氨丙基三甲氧基矽烷、2-氨乙基-氨丙基三甲氧基矽烷、氨乙基氨丙基甲基二甲氧基矽烷、氨乙基氨丙基甲基二乙氧基矽烷;
本發明的又一個目的是將上述網絡結構成炭劑應用於聚丙烯、聚乙烯以及乙烯-醋酸乙烯共聚物材料中,得到一種阻燃效果更好的高分子組合物;
本發明為了實現以上目的,採用以下技術方案:
成炭劑與聚磷酸鹽按比例充分混合均勻,得到復配後的阻燃劑;復配後的阻燃劑與聚合物在擠出機中通過熔融共混,得到具有優異阻燃性能和高熱穩定性能的高分子組合物。
本發明所述的聚合物為聚丙烯、聚乙烯、乙烯-醋酸乙烯酯共聚物。
本發明所述的聚磷酸鹽為聚磷酸銨、聚磷酸三聚氰胺中的一種。
本發明所述的成炭劑與聚磷酸鹽的重量比為1:5~3:1。
本發明所述的復配後的阻燃劑與聚合物的質量比為:10~50:100。
本發明所述的高分子組合物中也包括一般常見的助劑譬如穩定劑、分散劑、成核劑以及其它無機填料譬如雲母石、碳酸鈣、矽石或他們的混合物也可包括在內,所有成分加起來為100%重量百分比。
與現有技術相比,本發明的有益效果體現在以下方面:
本發明所製備的成炭劑,具有網絡結構,不溶不融,具有優異的耐水性,並且經過矽烷偶聯劑修飾後,成炭劑在高分子材料中具有更好的相容性和分散性,克服了小分子成炭劑易遷移、易吸水與基體相容性差的不足。另外,該成炭劑成炭效率高、合成工藝簡單,反應產率高,阻燃效果好。
附圖說明
圖1是本發明網絡結構成炭劑的紅外譜圖;
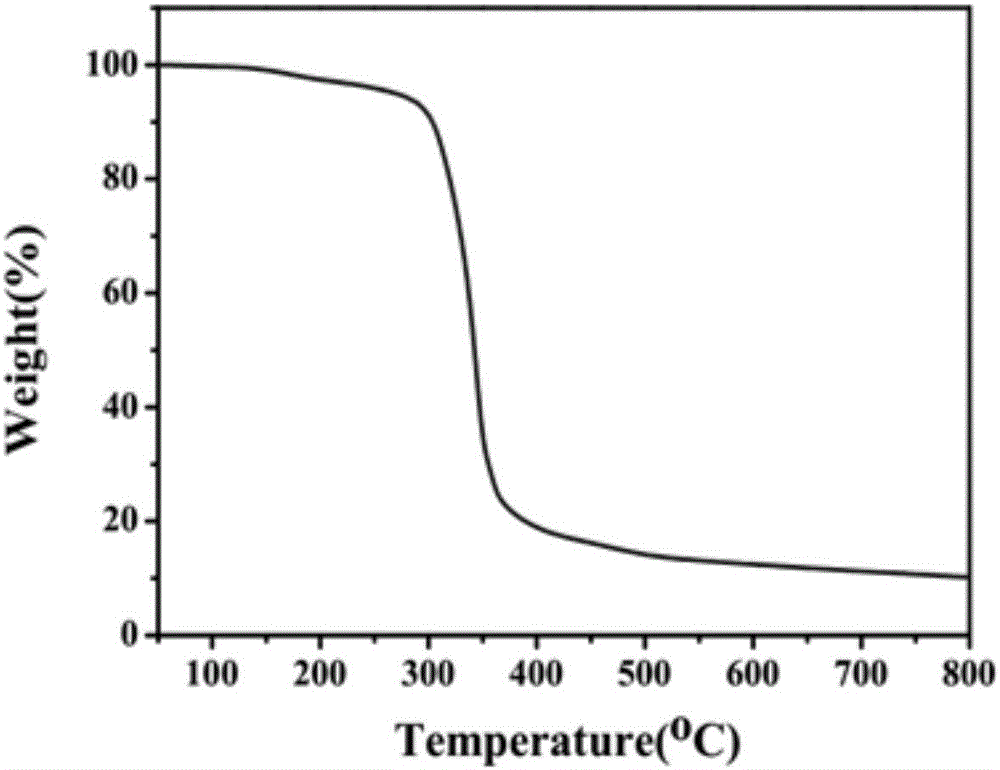
圖2是本發明網絡結構成炭劑的熱重圖。
具體實施方式
以下以具體的實施實例來說明本發明的技術方案,但本發明的保護範圍不限於此:
燃燒測試標準:GB/T 2408—2008;
氧指數測試標準:GB/T 2406.2—2006。
實施例1:網絡結構成炭劑的製備
稱取甲醛溶液0.20mol(16.23g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑N』N-二甲基甲醯胺120ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在50℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約0.5h滴加完畢,在50℃保溫反應2h得到澄清透明的溶液,之後向溶液中加入8%的三聚氰胺(1.01g),於50℃反應1h,繼續升溫至90℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應2h,得到白色固液混合物,反應結束後向溶液中加入0.5%(0.17g)的氨丙基三甲氧基矽烷偶聯劑進行功能化處理1h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率90%。在上述實驗過程中,哌嗪和所製備的含哌嗪結構的成炭劑的紅外譜圖分別如圖1a,b所示,圖1(a)中3209cm-1和1551cm-1為N-H的振動吸收峰,2855cm-1、1430cm-1為-C-H的振動吸收峰,1272為C-N基團振動峰吸收峰;而在圖1(b)中3209cm-1處N-H振動峰消失,並且出現了1514cm-1處的三個小峰為三聚氰胺碳氮骨架的震動吸收,由此可判斷成功合成了含哌嗪結構的成炭劑。所製備的成炭劑的熱重曲線如圖2所示,從圖中可以看出成炭劑具有良好的熱穩定性,能夠滿足高分子材料的加工溫度。
實施例2:網絡結構成炭劑的製備
稱取甲醛溶液0.20mol(16.23g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑N』N-二甲基乙醯胺120ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在20℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.63g,蒸餾水20ml),約0.5h滴加完畢,在20℃保溫反應10h得到澄清透明的溶液,之後向溶液中加入1%的三聚氰胺(0.13g),於20℃反應2h,繼續升溫至90℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.63g,蒸餾水20ml),滴畢後保溫反應4h,得到白色固液混合物,反應結束後向溶液中加入0.5%(0.17g)的氨丙基三甲氧基矽烷偶聯劑進行功能化處理1h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率85%。
實施例3:網絡結構成炭劑的製備
稱取乙醛0.30mol(13.22g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑二氧六環180ml,用三乙胺調節溶液PH在8-9,控制溶液溫度在30℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約0.5h滴加完畢,在30℃保溫反應8h得到澄清透明的溶液,之後向溶液中加入8%的三聚氰胺(1.00g),於30℃反應1.5h,繼續升溫至60℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應5h,得到白色固液混合物,反應結束後向溶液中加入1%(0.31g)的氨丙基三甲氧基矽烷偶聯劑進行功能化處理2h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率90%。
實施例4:網絡結構成炭劑的製備
稱取固體甲醛0.40mol(12.01g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑乙腈240ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在45℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.16mol,13.78g,蒸餾水30ml),約1h滴加完畢,在45℃保溫反應8h得到澄清透明的溶液,之後向溶液中加入10%的三聚氰胺(2.02g),於45℃反應2h,繼續升溫至90℃,向反應液中滴加哌嗪溶液(哌嗪0.16mol,13.78g,蒸餾水30ml),滴畢後保溫反應2h,得到白色固液混合物,反應結束後向溶液中加入0.8%(0.32g)的氨丙基三乙氧基矽烷偶聯劑進行功能化處理1h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率86%。
實施例5:網絡結構成炭劑的製備
稱取苯甲醛0.40mol(42.45g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑四氫呋喃240ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在0℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.16mol,13.78g,蒸餾水30ml),約1h滴加完畢,在0℃保溫反應10h得到澄清透明的溶液,之後向溶液中加入10%的三聚氰胺(2.02g),於0℃反應5h,繼續升溫至80℃,向反應液中滴加哌嗪溶液(哌嗪0.16mol,13.78g,蒸餾水30ml),滴畢後保溫反應2h,得到白色固液混合物,反應結束後向溶液中加入0.8%(0.92g)的2-氨乙基-氨丙基三甲氧基矽烷偶聯劑進行功能化處理2h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率80%。
實施例6:網絡結構成炭劑的製備
稱取甲醛溶液0.20mol(16.23g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑N』N-二甲基甲醯胺120ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在60℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約0.5h滴加完畢,在60℃保溫反應2h得到澄清透明的溶液,之後向溶液中加入4%的三聚氰胺(0.50g),於60℃反應1h,繼續升溫至70℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應3h,得到白色固液混合物,反應結束後向溶液中加入0.8%(0.26g)的2-氨乙基-氨丙基三甲氧基矽烷偶聯劑進行功能化處理1h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率83%。
實施例7:網絡結構成炭劑的製備
稱取糠醛0.20mol(19.22g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑二氧六環120ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在50℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約0.5h滴加完畢,在50℃保溫反應2h得到澄清透明的溶液,之後向溶液中加入1%的三聚氰胺(0.13g),於50℃反應1h,繼續升溫至80℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應2.5h,得到白色固液混合物,反應結束後向溶液中加入0.5%(0.19g)的氨丙基三甲氧基矽烷偶聯劑進行功能化處理1.5h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率78%。
實施例8:網絡結構成炭劑的製備
稱取固體甲醛0.30mol(9.01g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑乙腈180ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在50℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約1h滴加完畢,在50℃保溫反應2h得到澄清透明的溶液,之後向溶液中加入6%的三聚氰胺(0.76g),於50℃反應1h,繼續升溫至60℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應5h,得到白色固液混合物,反應結束後向溶液中加入0.5%(0.14g)的氨乙基氨丙基甲基二甲氧基矽烷偶聯劑進行功能化處理1h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率81%。
實施例9:網絡結構成炭劑的製備
稱取1-丁醛0.30mol(21.63g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑N』N-二甲基乙醯胺180ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在40℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約0.5h滴加完畢,在40℃保溫反應6h得到澄清透明的溶液,之後向溶液中加入3%的三聚氰胺(0.38g),於40℃反應2h,繼續升溫至60℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應5h,得到白色固液混合物,反應結束後向溶液中加入0.5%(0.20g)的氨乙基氨丙基甲基二甲氧基矽烷偶聯劑進行功能化處理1h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率79%。
實施例10:網絡結構成炭劑的製備
稱取甲醛溶液0.20mol(16.23g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑N』N-二甲基甲醯胺120ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在50℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約1h滴加完畢,在50℃保溫反應8h得到澄清透明的溶液,之後向溶液中加入8%的三聚氰胺(1.00g),於50℃反應2h,繼續升溫至85℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應3h,得到白色固液混合物,反應結束後向溶液中加入0.8%(0.28g)的氨丙基三甲氧基矽烷偶聯劑進行功能化處理2h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率83%。
實施例11:網絡結構成炭劑的製備
稱取苯甲醛0.20mol(21.22g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑乙腈120ml,用碳酸鉀調節溶液PH在8-9,控制溶液溫度在45℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約0.5h滴加完畢,在45℃保溫反應4h得到澄清透明的溶液,之後向溶液中加入8%的三聚氰胺(1.01g),於45℃反應3h,繼續升溫至60℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應5h,得到白色固液混合物,反應結束後向溶液中加入0.8%(0.32g)的氨乙基氨丙基甲基二乙氧基矽烷偶聯劑進行功能化處理1h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率80%。
實施例12:網絡結構成炭劑的製備
稱取固體甲醛0.20mol(6.01g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑四氫呋喃120ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在50℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約0.5h滴加完畢,在50℃保溫反應4h得到澄清透明的溶液,之後向溶液中加入8%的三聚氰胺(1.01g),於50℃反應2h,繼續升溫至90℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應2h,得到白色固液混合物,反應結束後向溶液中加入1%(0.24g)的氨丙基三甲氧基矽烷偶聯劑進行功能化處理2h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率88%。
實施例13:網絡結構成炭劑的製備
稱取乙醛0.30mol(13.21g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑N』N-二甲基甲醯胺180ml,用氫氧化鈉調節溶液PH在8-9,控制溶液溫度在25℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約0.5h滴加完畢,在25℃保溫反應8h得到澄清透明的溶液,之後向溶液中加入10%的三聚氰胺(1.26g),於25℃反應2h,繼續升溫至75℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應4h,得到白色固液混合物,反應結束後向溶液中加入0.8%(0.25g)的氨乙基氨丙基甲基二乙氧基矽烷偶聯劑進行功能化處理1.5h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率87%。
實施例14:網絡結構成炭劑的製備
稱取固體甲醛0.30mol(9.01g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑二氧六環180ml,用碳酸鈉調節溶液PH在8-9,控制溶液溫度在50℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),約0.5h滴加完畢,在50℃保溫反應5h得到澄清透明的溶液,之後向溶液中加入8%的三聚氰胺(1.01g),於50℃反應1h,繼續升溫至60℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應3.5h,得到白色固液混合物,反應結束後向溶液中加入0.8%(0.22g)的氨丙基三甲氧基矽烷偶聯劑進行功能化處理2h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率76%。
實施例15:網絡結構成炭劑的製備
稱取甲醛溶液0.20mol(16.23g),加入到裝有冷凝回流裝置的500ml三口瓶中,並加入有機溶劑乙腈120ml,用氫氧化鉀調節溶液PH在8-9,控制溶液溫度在50℃,於此溫度下開始滴加哌嗪水溶液(哌嗪0.10mol,8.16g,蒸餾水20ml),約0.5h滴加完畢,在50℃保溫反應4h得到澄清透明的溶液,之後向溶液中加入5%的三聚氰胺(0.63g),於50℃反應1h,繼續升溫至90℃,向反應液中滴加哌嗪溶液(哌嗪0.10mol,8.62g,蒸餾水20ml),滴畢後保溫反應2h,得到白色固液混合物,反應結束後向溶液中加入0.8%(0.22g)的氨丙基三甲氧基矽烷偶聯劑進行功能化處理1h,停止反應,降至室溫,抽濾,烘乾,得到目標產物成炭劑,產率87%。
實施例1-15得到的目標產物成炭劑分子結構如式(1):
式中1<n<10000;R的結構為下列之一:R1為含單個羥基或氨基的矽烷偶聯劑基團。
實施例16網絡結構成炭劑的應用
R為網絡結構成炭劑、多聚磷酸銨,按重量比1:3混合均勻,得到復配阻燃劑與聚丙烯按重量比10:100進行預混合,然後通過熔融擠出獲得阻燃複合物,按標準進行阻燃測試,UL94垂直燃燒為V-0級,極限氧指數LOI為25.2%。
實施例17網絡結構成炭劑的應用
R為的網絡結構成炭劑、多聚磷酸銨,按重量比2:1混合均勻,得到復配阻燃劑與聚丙烯按重量比30:100進行預混合,然後通過熔融擠出獲得阻燃複合物,按標準進行阻燃測試,UL94垂直燃燒為V-0級,極限氧指數LOI為26.2%。
實施例18網絡結構成炭劑的應用
R為網絡結構成炭劑、多聚磷酸三聚氰胺,按重量比1:5混合均勻,得到復配阻燃劑與聚乙烯按重量比45:100進行預混合,然後通過熔融擠出獲得阻燃複合物,按標準進行阻燃測試,UL94垂直燃燒為V-0級,極限氧指數LOI為28.4%。
實施例19網絡結構成炭劑的應用
R為的網絡結構成炭劑、多聚磷酸銨,按重量比3:1混合均勻,得到復配阻燃劑與聚乙烯按重量比30:100進行預混合,然後通過熔融擠出獲得阻燃複合物,按標準進行阻燃測試,UL94垂直燃燒為V-0級,極限氧指數LOI為26.8%。
實施例20網絡結構成炭劑的應用
R為的網絡結構成炭劑、多聚磷酸銨,按重量比1:2混合均勻,得到復配阻燃劑與乙烯-醋酸乙烯酯共聚物按重量比25:100進行預混合,然後通過熔融擠出獲得阻燃複合物,按標準進行阻燃測試,UL94垂直燃燒為V-0級,極限氧指數LOI為25.9%。
實施例21網絡結構成炭劑的應用
R為的網絡結構成炭劑、多聚磷酸三聚氰胺,按重量比1:3混合均勻,得到復配阻燃劑與乙烯-醋酸乙烯酯共聚物按重量比20:100進行預混合,然後通過熔融擠出獲得阻燃複合物,按標準進行阻燃測試,UL94垂直燃燒為V-0級,極限氧指數LOI為24.8%。
實施例22網絡結構成炭劑的應用
R為的網絡結構成炭劑、多聚磷酸銨,按重量比3:1混合均勻,得到復配阻燃劑與乙烯-醋酸乙烯酯共聚物按重量比50:100進行預混合,然後通過熔融擠出獲得阻燃複合物,按標準進行阻燃測試,UL94垂直燃燒為V-0級,極限氧指數LOI為31.2%。
實施例23網絡結構成炭劑的應用
R為的網絡結構成炭劑、多聚磷酸三聚氰胺,按重量比3:1混合均勻,得到復配阻燃劑與聚丙烯按重量比35:100進行預混合,然後通過熔融擠出獲得阻燃複合物,按標準進行阻燃測試,UL94垂直燃燒為V-0級,極限氧指數LOI為26.4%。
實施例24網絡結構成炭劑的應用
R為的網絡結構成炭劑、多聚磷酸銨,按重量比2:1混合均勻,得到復配阻燃劑與聚乙烯按重量比50:100進行預混合,然後通過熔融擠出獲得阻燃複合物,按標準進行阻燃測試,UL94垂直燃燒為V-0級,極限氧指數LOI為30.2%。
實施例25網絡結構成炭劑的應用
R為的網絡結構成炭劑、多聚磷酸三聚氰胺,按重量比1:2.5混合均勻,得到復配阻燃劑與乙烯-醋酸乙烯酯共聚物按重量比35:100進行預混合,然後通過熔融擠出獲得阻燃複合物,按標準進行阻燃測試,UL94垂直燃燒為V-0級,極限氧指數LOI為26.7%。
上述實施例並非是對於本發明的限制,本發明並非僅限於上述實施例,只要符合本發明要求,均屬於本發明的保護範圍。