一種右旋硫辛酸或其氨丁三醇鹽的雜質和製備方法以及它們的檢測方法與流程
2023-04-27 03:57:12 5
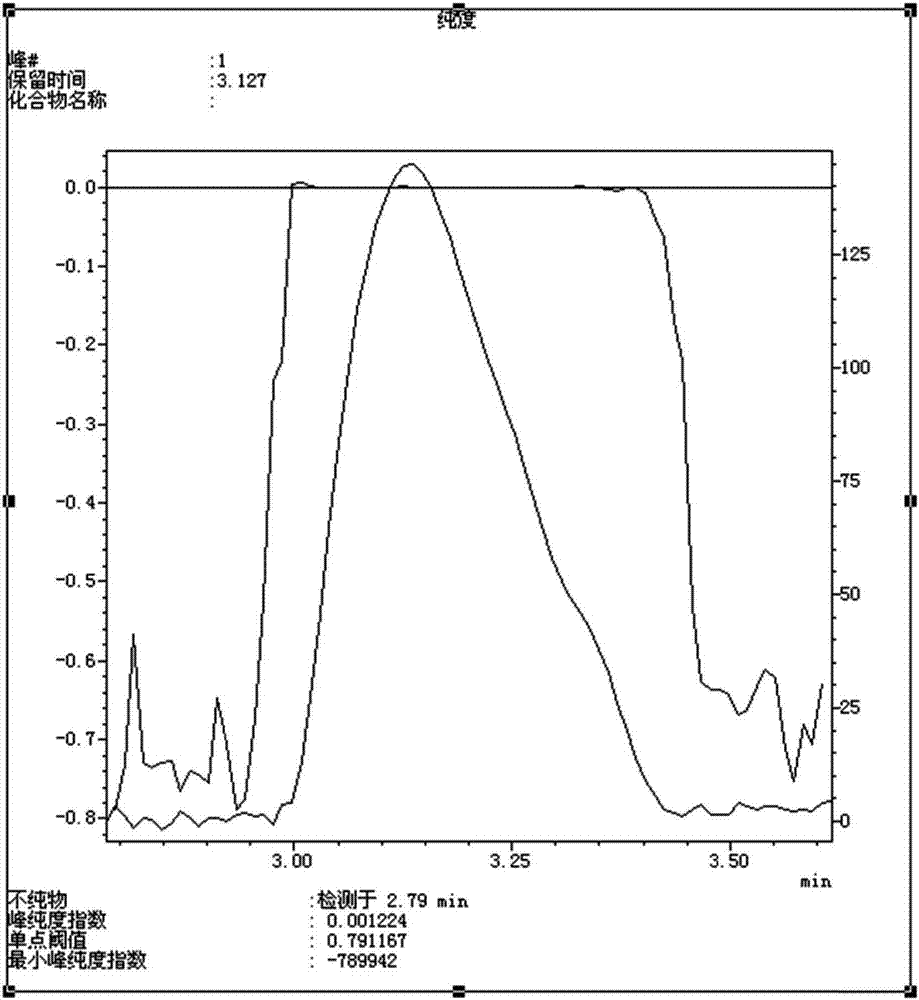
本發明涉及藥物化學和分析領域,具體涉及一種右旋硫辛酸或其氨丁三醇鹽的雜質化合物、其製備方法以及右旋硫辛酸或其氨丁三醇鹽有關物質的分析檢測方法。
背景技術:
:α-硫辛酸(α-lipoicacid,結構式如下),化學名為1,2-二硫戊環-3-戊酸,英文名l,2-dithiolane-3-pentanoicacid,具有優異的抗氧化作用,被譽為萬能抗氧化劑。硫辛酸在臨床上應用廣泛,可用於治療肝功能障礙、亞急性壞死腦病、神經性疾病、放射性傷害症、砷鎘汞等重金屬中毒等疾病的治療。隨著α-硫辛酸的藥理藥效學的深入研究,發現硫辛酸的兩個對映體顯示出不同的生物活性,其中r體生物活性遠高於s體,其在抗hiv病毒和抗腫瘤方面也有顯著的療效。但r-(+)-硫辛酸的熱穩定性差,在有氧條件下易聚合,不溶於水等現象,這些都影響了它的儲存和生物利用度。因此,通常將r-(+)-硫辛酸製成金屬鹽或有機鹼的鹽來增加它穩定性、提高其溶解度和生物利用度,以滿足醫藥市場需要。右旋硫辛酸氨丁三醇為α-硫辛酸消旋體進行拆分後,與氨基丁三醇成鹽所得的化合物,可用於製備注射劑。然而α-硫辛酸極易氧化產生雜質,嚴重影響產品使用的安全性,為了保障臨床用藥安全,對樣品中的雜質嚴格限制尤其重要。目前,α-硫辛酸質量標準已被美國藥典和歐洲藥典收錄,對有關物質檢測的色譜條件一致,但僅涉及部分已知雜質。文獻(oxidationofα-lipoicacid,j.org.chem.,vol.40,no.1,1975)公開了多種α-硫辛酸的氧化降解產物。cn105254612a也公開了硫辛酸的兩種氧化雜質。但本發明人深入研究發現,仍有部分雜質屬於未知雜質,尚未分離確證。技術實現要素:本發明的目的是提供新的右旋硫辛酸或其氨丁三醇鹽的雜質化合物、其製備方法和用途, 以及右旋硫辛酸或其氨丁三醇鹽有關物質的分析檢測方法,為右旋硫辛酸或其氨丁三醇鹽的原料及製劑的生產和質量研究提供了保障。本發明的一個方面是提供一種右旋硫辛酸或其氨丁三醇鹽的新的雜質化合物。本發明的第二方面是提供一種製備上述式(i)雜質化合物的方法。本發明的第三方面是提供上述式(i)雜質化合物的用途。本發明的第四方面是提供一種右旋硫辛酸或其氨丁三醇鹽有關物質的檢測方法。為此,本發明提供以下技術方案:根據本發明的第一方面,提供一種右旋硫辛酸或其氨丁三醇鹽的雜質化合物,其結構如式(i)所示,式(i)化合物化學名為(r)-5-(1,2-二硫戊烷-3-基)戊酸-2,2-二氧化物。所述雜質化合物可以作為雜質對照品使用,例如在右旋硫辛酸或其氨丁三醇原料或製劑的有關物質檢查中作為雜質對照品。根據本發明的第二方面,提供一種製備上述式(i)化合物的方法,包括步驟:1)將右旋硫辛酸氨丁三醇用純化水溶解,配製成0.01~1g/ml的水溶液,用光照射該水溶液,得到一次製備樣品溶液;例如配製成0.06g/ml的水溶液,用4500lx±500lx強光照射水溶液15天;2)將上述樣品溶液經過高效液相色譜(hplc)分離製備得到式(i)所示的雜質化合物,所述hplc製備採用二次製備法,其中:一次製備的條件是:固定相:十八烷基矽烷鍵合矽膠流動相:a:0.15%磷酸水溶液,b:乙腈檢測波長:215nm流速:200ml/min柱溫:室溫洗脫方式:梯度洗脫收集保留時間約50-60min處的目標峰對應的物質。將一次製備收集的目標物質進行二次製備,二次製備的條件是:固定相:十八烷基矽烷鍵合矽膠流動相:a:0.1%的三氟乙酸水溶液,b:乙腈檢測波長:215nm流速:200ml/min柱溫:室溫洗脫方式:梯度洗脫收集保留時間約31-33min處的目標峰對應的物質。根據本發明的第三方面,提供式(i)所示的化合物在右旋硫辛酸或其鹽(例如氨丁三醇鹽)及其製劑的有關物質的檢查中作為雜質對照品的用途。所述製劑可以是注射劑或固體製劑,所述固體製劑可以是片劑或膠囊等。根據本發明的第四方面,提供一種右旋硫辛酸或其氨丁三醇鹽的有關物質的檢測方法,所述方法為hplc法,檢測條件為:固定相:十八烷基矽烷鍵合矽膠流動相:a:0.02%磷酸溶液,b:0.02%磷酸溶液-乙腈(40:60)檢測波長:215nm柱溫:室溫流速:1.0ml/min梯度洗脫:本發明提供了一種新的右旋硫辛酸或其氨丁三醇鹽的雜質化合物及其製備方法,該雜質可以作為雜質對照品,用於右旋硫辛酸或其氨丁三醇鹽及其製劑的質量研究。此外還提供了右旋硫辛酸或其氨丁三醇鹽的有關物質的檢測方法,可以有效分離檢測右旋硫辛酸或其氨丁三醇鹽及其多種氧化雜質、光照雜質等的含量,為右旋硫辛酸或其氨丁三醇鹽及製劑的質量研究提供了更準確和適用的分析方法。附圖說明圖1:式(i)化合物的1h-nmr核磁共振譜圖;圖2:根據usp收錄的方法進行的右旋硫辛酸氨丁三醇不同降解樣品的分析檢測圖;圖3:根據usp收錄的方法進行的氧化降解產物的峰純度檢測圖;圖4:根據本發明方法進行的右旋硫辛酸氨丁三醇不同降解樣品的分析檢測圖;圖5:根據本發明方法進行的右旋硫辛酸氨丁三醇有關雜質的分析檢測圖。圖6:根據本發明方法進行的式(i)雜質化合物的對照品定位分析檢測圖。具體實施方式以下實施例僅是舉例說明的作用,以便技術人員更好地理解本發明的優點,而不以任何方式限制本發明所揭示的內容。所採用的儀器和試劑均為本領域普通市售產品。本發明hplc檢測的柱溫為室溫,優選25-35℃。未特別說明的步驟均為本領域的常規操作,例如進樣前先平衡柱子等。實施例1:(r)-5-(1,2-二硫戊烷-3-基)戊酸-2,2-二氧化物(式i化合物)的製備將60g右旋硫辛酸氨丁三醇溶解在1000ml純化水中,配製成0.06g/ml的溶液,以4500lx±500lx強光照射水溶液15天,得到一次製備樣品溶液。hplc一次製備的色譜條件:儀器:創新通恆製備液相lc6000,製備柱dac100,直徑10cm固定相:十八烷基矽烷鍵合矽膠,1.5kg裝料量,10μm流動相:a-0.15%磷酸水溶液,b-乙腈檢測波長:215nm流速:200ml/min柱溫:25℃進樣量:200ml洗脫方式:梯度洗脫收集保留時間約50-60min處的目標峰對應的物質。該粗品純化過程中,目標峰是在約53-54min處的較寬的峰,呈小包狀,收集時可選擇將50-60min段全部收集,再進行下一步純化。將一次製備收集的目標物質濃縮至一半體積,進行二次製備,hplc二次製備的色譜條件:儀器:創新通恆製備液相lc6000,製備柱dac100,直徑10cm固定相:十八烷基矽烷鍵合矽膠,1.5kg裝料量,10μm流動相:a:0.1%的三氟乙酸水溶液,b:乙腈檢測波長:215nm流速:200ml/min柱溫:25℃進樣量:200ml洗脫方式:梯度洗脫收集保留時間約32.1min處的目標峰對應的物質。將收集的目標物質濃縮後得到約136mg式(i)化合物。結構確證:1)質譜儀器:agilent6540bq-tof質譜儀,離子源:電噴霧,質量分析器:離子阱。測得供試品準分子的離子峰[m-h]-的m/z為237.01,是式(i)化合物(c8h16o4s2,mw=238.32)減一質子得到,質譜數據與式(i)化合物的分子量一致。2)1h-nmr核磁共振譜儀器:brukerdrx500,溶劑dmso-d6,內標tms,溫度303k。核磁共振氫譜測定數據如下:化學位移(ppm)質子數多重性歸屬12.0031s11位氧原子上的活潑氫3.621-2.8453m1位-ch2-上的質子,以及4位=ch-上的質子2.299-1.9743m9位-ch2-上的質子,以及5位-ch2-上的其中一個質子1.7591m5位-ch2-上的其中一個質子1.615-1.0866m6,7,8位-ch2-上的質子通過分析氫譜,可以判斷該化合物為(r)-5-(1,2-二硫戊烷-3-基)戊酸-2,2-二氧化物,即式(i)化合物。實施例2:根據usp收錄的方法分離檢測右旋硫辛酸氨丁三醇有關物質本實施例考察usp收錄的方法對右旋硫辛酸氨丁三醇樣品中主成份與有關物質的分離效果。右旋硫辛酸氨丁三醇的各樣品的製備:樣品1(未破壞):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,用流動相溶解並稀釋至刻度,搖勻。樣品2(氧化破壞):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,加3%過氧化氫溶液1ml溶解,立即用流動相溶解並稀釋至刻度,搖勻。樣品3(高溫破壞):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,加水1ml溶解後,於100℃水浴中加熱2.5h後,用流動相溶解並稀釋至刻度,搖勻。樣品4(光照破壞):取右旋硫辛酸氨丁三醇約15mg,精密稱定,加水1ml溶解,置10ml量瓶中,於4500lx±500lx照度下,放置18.5h,用流動相溶解並稀釋至刻度,搖勻。樣品5(酸破壞):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,加0.1mol/l鹽酸溶液1ml,室溫放置3.5h後,加入0.1mol/l氫氧化鈉溶液1ml中和,用流動相溶解並稀釋至刻度,搖勻。樣品6(鹼破壞):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,加1mol/l氫氧化鈉溶液1ml,室溫放置4h後,加入1mol/l鹽酸溶液1ml中和,用流動相溶解並稀釋至刻度,搖勻。檢測方法:hplc法,取各樣品溶液進樣,注入液相色譜儀,記錄色譜圖。檢測條件為:儀器:lc-20at高效液相色譜儀(日本島津,shimpackvp-ods,4.6mm×250mm,5μm)固定相:十八烷基矽烷鍵合矽膠流動相:甲醇-緩衝鹽(0.68g/l磷酸二氫鉀溶液)-乙腈(58:46:9),用磷酸溶液(8.3→100)調節ph值至3.0~3.1檢測波長:215nm柱溫:35℃流速:1.2ml/min進樣量:20μl檢測圖譜:見圖2、圖3。從圖2可以看出:右旋硫辛酸氨丁三醇極易被氧化;且在其他降解條件下,與氧化降解產物相同保留時間處,也存在其他降解雜質色譜峰;經峰純度考察(圖3),氧化降解產物峰純度指數為0.001224,小於0.998,提示此色譜峰不是單一色譜峰,該檢測方法無法將氧化雜質分離,不能準確的對單個氧化雜質進行定量,且不能用此方法對氧化雜質進行製備以用於 有關物質檢測方法學研究。實施例3:根據本發明的方法分離檢測右旋硫辛酸氨丁三醇有關物質本實施例考察本發明的方法對右旋硫辛酸氨丁三醇樣品中主成份與有關物質的分離效果。右旋硫辛酸氨丁三醇的各樣品的製備:樣品1(未破壞):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,用稀釋液[0.05%磷酸溶液-乙腈(85:15)]溶解並稀釋至刻度,搖勻。樣品2(氧化破壞):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,加3%過氧化氫溶液1ml溶解,立即加稀釋液[0.05%磷酸溶液-乙腈(85:15)]稀釋至刻度,搖勻。樣品3a(高溫破壞-固態):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,於100℃水浴中加熱2h後,用稀釋液[0.05%磷酸溶液-乙腈(85:15)]溶解並稀釋至刻度,搖勻。樣品3b(高溫破壞-液態):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,加水1ml溶解後,於100℃水浴中加熱2h後,用稀釋液[0.05%磷酸溶液-乙腈(85:15)]稀釋至刻度,搖勻。樣品4a(光照破壞-固態):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,於4500lx±500lx照度下,放置24h,用稀釋液[0.05%磷酸溶液-乙腈(85:15)]溶解並稀釋至刻度,搖勻。樣品4b(光照破壞-液態):取右旋硫辛酸氨丁三醇約15mg,精密稱定,加水1ml溶解,置10ml量瓶中,於4500lx±500lx照度下,放置24h,用稀釋液[0.05%磷酸溶液-乙腈(85:15)]稀釋至刻度,搖勻。樣品5(酸破壞):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,加0.1mol/l鹽酸溶液1ml,室溫放置8h後,加入0.1mol/l氫氧化鈉溶液1ml中和,用稀釋液[0.05%磷酸溶液-乙腈(85:15)]稀釋至刻度,搖勻。樣品6(鹼破壞):取右旋硫辛酸氨丁三醇約15mg,精密稱定,置10ml量瓶中,加1mol/l氫氧化鈉溶液1ml,室溫放置8h後,加入1mol/l鹽酸溶液1ml中和,用稀釋液[0.05%磷酸溶液-乙腈(85:15)]稀釋至刻度,搖勻。樣品7(鹼破壞-高溫):取本品約15mg,精密稱定,置10ml量瓶中,加1mol/l氫氧化鈉溶液1ml,於100℃水浴中加熱0.5h後,加入1mol/l鹽酸溶液1ml中和,用稀釋液[0.05%磷酸溶液-乙腈(85:15)]稀釋至刻度,搖勻。檢測方法:hplc法,取各樣品溶液進樣,注入液相色譜儀,記錄色譜圖。檢測條件為:儀器:lc-20at高效液相色譜儀(日本島津,shimpackvp-ods,4.6mm×250mm,5μm)固定相:十八烷基矽烷鍵合矽膠流動相:a:0.02%磷酸溶液,b:0.02%磷酸溶液-乙腈(40:60)檢測波長:215nm柱溫:25℃流速:1.0ml/min進樣量:20μl洗脫方式:梯度洗脫檢測圖譜:如圖4所示。從圖4可以看出,在本發明的色譜條件下,分離出4個氧化降解雜質的單峰,右旋硫辛酸氨丁三醇與降解雜質之間的分離度良好,各雜質之間也得到了有效分離。本發明的方法能夠有效分離檢測右旋硫辛酸氨丁三醇樣品中存在的降解雜質,尤其是能更定量分析四種主要的氧化雜質,可作為右旋硫辛酸氨丁三醇有關物質的檢測方法。實施例4:根據本發明的方法檢測右旋硫辛酸氨丁三醇有關物質的含量本實施例示範性地說明本發明的方法用於檢測右旋硫辛酸氨丁三醇有關物質的含量。1、供試品溶液製備取右旋硫辛酸氨丁三醇約15.02mg,精密稱定,置10ml量瓶中,加3%過氧化氫溶液1ml溶解,立即加稀釋液[0.05%磷酸溶液-乙腈(85:15)]稀釋至刻度,搖勻。2、光照降解雜質(式(i)化合物)對照品溶液的製備取光照降解雜質約1mg,用甲醇溶解並稀釋製成每1ml中約含1mg的溶液,搖勻。3、檢測操作檢測方法:hplc法,取供試品溶液20μl,注入液相色譜儀,記錄色譜圖。檢測條件為:儀器:lc-20at高效液相色譜儀(日本島津,shimpackvp-ods,4.6mm×250mm,5μm)固定相:十八烷基矽烷鍵合矽膠流動相:a:0.02%磷酸溶液,b:0.02%磷酸溶液-乙腈(40:60)檢測波長:215nm柱溫:25℃流速:1.0ml/min進樣量:20μl洗脫方式:梯度洗脫檢測結果:圖譜如圖5、圖6所示。結果分析:色譜峰1、2、3、4為氧化降解雜質峰,分別為22.716、26.337、28.542、29.619分鐘,與實施例3是一致的,相對主峰的保留時間分別為0.59、068、0.74和0.76,峰面積百分比分別為3.058%、6.748%、8.320%、6.648%;色譜峰6為右旋硫辛酸氨丁三醇峰,保留時間為38.798分鐘;色譜峰7為光照降解雜質(式(i)化合物)峰,保留時間為44.360分鐘,相對主峰的保留時間分別為1.14,峰面積百分比為0.043%;光照降解雜質(式(i)化合物)峰對照品色譜峰保留時間為44.093分鐘,與色譜峰7基本一致;色譜峰5為未知雜質。從上述實施例可以看出,本發明方法的檢測條件優於usp收錄的檢測方法,能夠有效分離檢測右旋硫辛酸或其氨丁三醇鹽的有關物質的含量,尤其是定量檢測四個氧化降解雜質和光照降解雜質。相比現有技術方法,本發明的方法更適用於右旋硫辛酸或其氨丁三醇鹽和製劑的質量研究和控制。以上實施例僅是解釋說明的目的,本領域普通技術人員可以在本發明描述的基礎上進行 改變和修飾,例如基於本發明調整hplc檢測條件,如樣品溶液配製、進樣量、柱溫、流動相比例,保留時間等發生的較小變化均屬於本發明的構思,只要在不違背本發明的精神前提下做出的改變,均屬於本發明所揭示和保護的範圍。當前第1頁12