用於製備前列腺素醯胺的新的方法與流程
2024-03-27 04:21:05 5
技術領域
本發明的主題是用於製備通式(I)的前列腺素(prostaglandin)醯胺的方法。
在通式(I)的化合物中
取代基的含義如下:
用虛線標示的鍵可以是單鍵或雙鍵,在位置5,6及13,14為雙鍵的情形下,其可以是順式或反式取向,
Q表示羥基且Z表示羥基或氧代基團,
R1及R2獨立地表示氫原子或直鏈或支鏈的C1-10烷基或芳烷基,任選地經-ONO2、芳烷基或芳基取代,其含有雜原子,
R3表示直鏈或支鏈、飽和或不飽和的C4-6烴基,或C4-10烷基環烷基或環烷基,或任選地經烷基或滷素原子取代的苯基、C7-10烷基芳基或雜芳基,
Y表示(CH2)n或O原子或S原子,且
n=0-3。
背景技術:
為了經濟地製備前列腺素醯胺衍生物,適當經取代的前列腺酸(prostan acid)須經活化。
根據目前的技藝狀態,羧酸可以通過轉化成其混合的酐、活化的酯或活化的醯胺而經活化,而且這些化合物可以隨後通過與適當的胺反應而進一步轉化成所要的前列腺素醯胺衍生物。
對於上述的可能性,化學性非常敏感的前列腺素酸通過酯形成的活化作用描述於例如EP 0 660 716中。
根據此方法,藉助於烷基滷化物而形成起始的酯且該酯隨後與適當的胺反應而得到醯胺官能團。此方法的缺點是要避免在合成結束時(即最後步驟)使用烷基滷化物,因為烷基滷化物經證明是具有遺傳毒性的藥物。此外,所得的酯必須在高溫與適當的胺長時間反應且轉化很少超過50%(EP0660716,42頁,實施例12)。考慮前列腺素的已知的溫度敏感性,其在高溫的處理不利地影響以此方式所得的前列腺素衍生物的雜質分布及收率。
混合的酐的製備及其與經適當取代的胺的反應陳述於WO9153206中。
此方法的缺點是用於製備混合的酐的活性烷化劑-滷化的甲酸酯、特戊醯氯及其它-經證明是具有遺傳毒性的化合物。
在WO2005058812(23頁)陳述的方法中,起始的羧酸通過使用活化劑1-(3-二甲基氨基丙基)-3-碳二亞胺鹽酸鹽(EDC HCl)及乙胺而直接轉化成乙基醯胺。在醯胺化反應期間,在位置11及15的羥基用四氫吡喃(THP)保護基保護,其隨後被移除。
技術實現要素:
根據本發明,我們發現通過新的經活化的酯及新的經活化的醯胺,可以在溫和的反應條件下,以高收率及高純度製備通式(I)的化合物。
根據本發明,製備通式(I)的化合物可以通過使通式(II)的酸:
其中在式中
用虛線標示的鍵可以是單鍵或雙鍵,在位置5,6及13,14為雙鍵的情形下,其可以是順式或反式取向,
Q表示羥基且Z表示羥基或氧代基團,
R3表示直鏈或支鏈、飽和或不飽和的C4-6烴基,或C4-10烷基環烷基或環烷基,或任選地經烷基或滷素原子取代的苯基、C7-10烷基芳基或雜芳基,
Y表示(CH2)n基團或O原子或S原子,且
n=0-3,
i)與合適引入R4的化合物反應,其中R4表示式a.)的基團,
a.)
且使得如此所得的通式(III)的醯胺:
其中Q、Z、R3、R4、Y及n的含義如上述定義,
與通式(IV)的胺反應
NHR1R2 IV.
NHR1R2 IV.
其中R1及R2的含義如上述定義,或
ii)與合適引入R5的化合物反應,其中R5表示式b.)、c.)、d.)或e.)的基團,
其中(X)表示滷素原子或氫原子,
且使得如此所得的通式(V)的經活化的酯:
其中Q、Z、R3、R5、Y及n的含義如上述定義,
與通式(IV)的胺反應,其中R1及R2的含義如上述定義。
我們還發現根據本發明通式(I)的化合物也可以通過在2-氯-1,3-二甲基咪唑啉鎓氯化物及鹼存在下,使通式(II)的化合物與通式(IV)的化合物反應而製備,其中在式中的取代基的含義如上述定義(方法iii)。
通式(III)及(V)的中間體是新的化合物。
對於合適引入R4的化合物,優選可使用1,1』-羰基二咪唑(DCI)或1,1』-硫羰基二咪唑,對於在存在活化劑的給定情況下引入R5的化合物,可使用N-羥基琥珀醯亞胺、N-羥基鄰苯二甲醯亞胺、N-羥基-5-降冰片烯-內-2,3-二甲醯胺、1-羥基苯並三唑、(苯並三唑-1-基氧基)三(二甲基氨基)鏻六氟磷酸鹽、N,N』-二琥珀醯亞胺基碳酸酯(DSC)或N,N』-二琥珀醯亞胺基草酸酯,特別是N,N』-二琥珀醯亞胺基碳酸酯。
對於活化劑,可使用N,N』-二異丙基碳二亞胺、N,N』-二環己基碳二亞胺或2-氯-1,3-二甲基咪唑啉鎓氯化物,優選N,N』-二異丙基碳二亞胺。
根據本發明在方法i)的過程中,可以在醚類溶劑或芳族溶劑或極性非質子溶劑或在它們的混合物中引入R4,使用例如二異丙醚、叔丁基甲基醚、2-甲基四氫呋喃、甲苯、茴香醚、二甲基甲醯胺、二甲亞碸、N-甲基吡咯烷酮,特別是四氫呋喃。所得的通式(III)經活化的醯胺在分離後或無分離時與通式(IV)的胺反應。引入基團R4期間的反應溫度是在20-80℃之間,優選是70℃,而式(III)與(IV)化合物的反應是在20-80℃之間進行,優選在室溫進行。
根據本發明在方法ii)的過程中,引入基團R5是在醚類溶劑或芳族溶劑或極性非質子溶劑或在它們的混合物中進行,使用例如二異丙醚、叔丁基甲基醚、2-甲基四氫呋喃、甲苯、茴香醚、二甲基甲醯胺、二甲亞碸、N-甲基吡咯烷酮,特別是四氫呋喃。所得的通式(V)經活化的酯在分離後或無分離時與通式(IV)的胺反應。引入基團R5期間的反應溫度是在0-80℃之間,優選在室溫,而式(V)與(IV)化合物的反應是在20-80℃之間進行,優選在室溫進行。
根據本發明在方法iii)的過程中,反應是在醚類溶劑或芳族溶劑或極性非質子溶劑或在它們的混合物中進行,使用例如二異丙醚、叔丁基甲基醚、2-甲基四氫呋喃、甲苯、茴香醚、二甲基甲醯胺、二甲亞碸、N-甲基吡咯烷酮或四氫呋喃。至於鹼,可以使用經常使用的鹼,例如吡啶、N-甲基嗎啉、二異丙基乙基胺、1,5-二氮雜二環[4,3,0]壬-5-烯、1,8-二氮雜二環[5,4,0]十一碳-7-烯或三乙胺。
反應是在0-70℃之間的溫度進行,其通過向通式(II)化合物在有機溶劑中的溶液中在0-70℃優選30℃加入通式(IV)化合物、2-氯-1,3-二甲基咪唑啉鎓氯化物及2摩爾當量的鹼。將混合物先在此溫度攪拌且隨後逐漸加熱直到起始物質消失。通過TLC追蹤反應。
方法i、ii或iii也可以在一鍋法條件下進行。
對於通式(IV)的胺,在比馬前列素的情形下,適於最終化合物的胺,可以使用乙胺。
對於通式(IA)化合物的製備
其中R1、R2、R3、Y及n的含義如上述定義,
使用根據本發明通式(IIA)的化合物作為起始物質。
通式(IIA)的化合物:
其中在式中
R3表示直鏈或支鏈、飽和或不飽和的C4-6烴基,或C4-10烷基環烷基或環烷基,或任選地經烷基或滷素原子取代的苯基、C7-10烷基芳基或雜芳基,
Y表示(CH2)n基團或O原子或S原子,且
n=0-3,
可以根據本發明製備,其通過將通式(XII)的內酯二醇(lactondiol):
其中R3、Y及n的含義如上述定義,
還原為通式(XIII)的內酯三醇(lactontriol):
其中R3、Y及n的含義如上述定義,
隨後移除式(XIII)化合物的保護基,
且將如此得到通式(XIV)的化合物,
其中R3、Y及n的含義如上述定義,
通過Wittig反應而轉化成通式(IIA)的化合物。
通式(XII)化合物的還原可以通過已知方法進行,例如用二異丁基氫化鋁在四氫呋喃介質中。保護基可以通過已知方法在酸性或鹼性介質中移除,優選在鹼性介質中移除。
通式(XIII)的內酯三醇衍生物:
其中R3表示直鏈或支鏈、飽和或不飽和的C4-6烴基,或C4-10烷基環烷基或環烷基,或任選地經烷基或滷素原子取代的苯基、C7-10烷基芳基或雜芳基,
Y表示(CH2)n基團或O原子或S原子,且n=0-3,
是新的化合物。
根據本發明的另一個具體實施方案,具體的通式IIA化合物式(IIB)化合物也可以製備成結晶形式,其中R3表示苯基且Y表示-(CH2)-基團。
(IIB)
式(IIB)化合物的結晶形式是新的化合物。
式(IIB)化合物可以以結晶形式製備,其通過向含有式(IIB)化合物的混合物中加入酯類及醚類溶劑的混合物。
根據此方法,使用二甲醚、乙醚、二異丙醚優選乙醚及二異丙醚作為醚型溶劑,及使用乙酸乙酯、乙酸甲酯、乙酸異丙酯優選乙酸異丙酯作為酯型溶劑。結晶作用是在-30℃及30℃之間進行,優選在0-25℃之間進行。將如此所得的結晶的混懸液攪拌1-24小時,優選8小時,隨後過濾並用醚類溶劑洗滌,優選用二異丙醚洗滌。過濾後的結晶在25-50℃之間真空乾燥,優選在35-40℃真空乾燥。
通式(II)及(XII)的化合物可以通過已知的方法製備,例如描述於US 5359095、WO 93/00329。
本發明方法的優點是所要的比馬前列素最終產物(如果需要時)可以通過結晶的比馬前列素酸合成。本發明方法的另一個優點是所要的最終產物是通過新的中間體、結晶、活化的酯或醯胺合成,其(如果需要時)可以分離且(如果需要時)可以通過結晶或色譜法純化。由於施加羧酸活化劑(例如DSC、DCI),不需要對位置9、11及15的仲羥基進行保護,沒有觀察到平行的反應例如二聚物形成,不論是在活化的酯或活化的醯胺的情形還是在最終醯胺形成的條件下,且本發明經活化的羧酸衍生物容易以高收率及純度分離。
令人驚奇地發現本發明結晶經活化的羧酸衍生物可以通過結晶法容易地純化以消除雜質且也可以在溫和的反應條件下且以高收率簡易地轉化成所要的醯胺最終產物。
熟知的是在活性藥物成分(API)的情形中,雜質的量是一個關鍵問題,在比馬前列素的情形中,每種未知雜質的量必須低於0.1%。為了保持此非常嚴格的限制,根據本發明的方法使用比馬前列素酸的結晶作用及活性羧酸衍生物的結晶作用代替在WO09153206中所述的昂貴的製備性HPLC拆分。
本發明的另一個實施方案是製備式(IB)的比馬前列素的高熔點結晶形式II的方法。
通過本發明的方法,可以製備化學與熱力學穩定的且無其它結晶形式的高熔點結晶形式II的比馬前列素。
下面的專利申請是涉及比馬前列素產物的結晶作用:US 2005/0209337 A1、WO 2009/153206 A2、US 2009/0163596 A1。
在US 2009/0163596 A1的實施例30中,比馬前列素的高熔點結晶形式(DSC峰讀取值為79℃)(進一步:結晶形式II)通過其X-射線衍射、其在KBr片中的IR光譜及其DSC及TGA曲線進行表徵。
WO 2009/153206描述了通過製備性HPLC、隨後通過結晶法而純化比馬前列素產物。結晶作用是由乙腈溶劑或從作為溶解溶劑的乙腈和作為沉澱溶劑的TBME(叔丁基甲基醚)進行。根據該描述,通過此方法可以製備比馬前列素的高熔點結晶形式(DSC峰讀取值為79℃)。通過重複該方法,申請人沒有成功得到比馬前列素的高熔點結晶形式。
US 2009/0163596描述了比馬前列素的結晶形式I及其製備。比馬前列素的結晶形式I通過其熔點(62-64℃)、DSC、X-射線衍射及IR研究而表徵。其詳細描述結晶方法,例如溶解方法(在接近沸點溫度的有機溶劑中或有機溶解溶劑與沉澱溶劑的混合物中)、冷卻方法、從母液分離沉澱的結晶形式及乾燥方法(在低溫的真空下)。比馬前列素的高熔點結晶形式(DSC峰讀取值為79℃)無法通過在US 2009/0163596所述的方法製備。
通過本發明的方法,可以製備比馬前列素的化學與熱力學穩定的、高熔點的結晶形式II(=高熔點的結晶形式(DSC峰讀取值為79℃),其不含結晶形式I。形式II通過其熔點(72-78℃)、DSC研究及IR與X-射線粉末衍射研究而表徵。
本發明方法的本質是,由在後處理且蒸發後含有比馬前列素的反應混合物,或由比馬前列素的任何結晶或非結晶的形式或由它們的任何比例的混合物,通過從質子或醚類溶劑的結晶作用,可以製備熱力學穩定的純的形式II。結晶方法如下:將計算量的溶劑添加至油性或結晶的粗比馬前列素中,然後乾燥並定期暴露至機械效應。
根據上述,本發明涉及用於製備式(IB)的比馬前列素的結晶形式II的方法,其特徵是將計算量的醚類或質子溶劑添加至在後處理且蒸發後的含有比馬前列素的反應混合物或比馬前列素的任何結晶或非結晶的形式或它們的任何比例的混合物中,如果需要時將所得的混合物暴露至機械效應,然後乾燥並均勻化。
在上述方法中所得的結晶形式II的熔點是在72-78℃之間,根據DSC研究的吸熱峰是在73-79℃之間且熔化熱高於75焦耳/克。
圖VI、VIII及IV分別呈現在根據本發明方法所得的結晶形式II的DSC曲線、X-射線粉末衍射曲線及IR光譜。
圖V、VII及III分別呈現結晶形式I的DSC曲線、X-射線粉末衍射曲線及IR光譜。
在根據本發明的方法中,申請人使用計算的量、適宜是20-60重量%、優選是35重量%的質子溶劑,特別是醇類例如甲醇、乙醇和/或水。優選使用水作為質子溶劑。
至於機械效應,申請人施加攪拌或刮擦,或兩者。添加的溶劑通過乾燥除去。乾燥在-60℃及70℃之間的溫度在真空進行,尤其在35℃在真空進行。
至於醚類溶劑,申請人施加計算的量、優選是2000-8000重量%的二甲醚、乙醚、二異丙醚,優選乙醚。添加的溶劑通過乾燥除去。乾燥是在低溫通過使得氮氣經過而進行,優選在0-(-)50℃之間通過使得氮氣經過而進行。
本發明還涉及以下方面:
項1.用於製備通式(I)的前列腺素醯胺的方法,
-其中在式中
用虛線標示的鍵可以是單鍵或雙鍵,在位置5,6及13,14為雙鍵的情形下,其可以是順式或反式取向,
Q表示羥基且Z表示羥基或氧代基團,
R1及R2獨立地表示氫原子或直鏈或支鏈的C1-10烷基或芳烷基,任選地經-ONO2、芳烷基或芳基取代,其含有雜原子,
R3表示直鏈或支鏈、飽和或不飽和的C4-6烴基,或C4-10烷基環烷基或環烷基,或任選地經烷基或滷素原子取代的苯基、任選地經烷基或滷素原子取代的C7-10烷基芳基或任選地經烷基或滷素原子取代的雜芳基,
Y表示(CH2)n基團或O原子或S原子,且其中n=0-3,
所述方法的特徵在於:
使得通式(II)的酸:
其中在式中
用虛線標示的鍵可以是單鍵或雙鍵,在位置5,6及13,14為雙鍵的情形下,其可以是順式或反式取向,
Q表示羥基且Z表示羥基或氧代基團,
R3表示直鏈或支鏈、飽和或不飽和的C4-6烴基,或C4-10烷基環烷基或環烷基,或任選地經烷基或滷素原子取代的苯基、任選地經烷基或滷素原子取代的C7-10烷基芳基或任選地經烷基或滷素原子取代的雜芳基,
Y表示(CH2)n基團或O原子或S原子,且其中n=0-3,
i)與合適引入基團R4的化合物反應,其中R4表示式a.)的基團,
且使得如此所得的通式(III)的醯胺
其中Q、Z、R3、R4、Y及n的含義如上述定義,
與通式(IV)的胺反應
NHR1R2 IV.
-其中R1及R2的含義如上述定義,或
ii)與合適引入基團R5的化合物反應,其中R5表示式b.)、c.)、d.)或e.)的基團,
其中(X)表示滷素原子或氫原子,
且使得如此所得的通式(V)的經活化的酯
其中Q、Z、R3、R5、Y及n的含義如上述定義,
與通式(IV)的胺反應,其中R1及R2的含義如上述定義,或
iii.)在2-氯-1,3-二甲基咪唑啉鎓氯化物及鹼存在下,與通式(IV)的胺反應,其中R1及R2的含義如上述定義。
項2.通式(III)的化合物,
其中在式中
用虛線標示的鍵可以是單鍵或雙鍵,在位置5,6及13,14的雙鍵的情形下,其可以是順式或反式取向,
Q表示羥基且Z表示羥基或氧代基團,
R3表示直鏈或支鏈、飽和或不飽和的C4-6烴基,或C4-10烷基環烷基或環烷基,或任選地經烷基或滷素原子取代的苯基、任選地經烷基或滷素原子取代的C7-10烷基芳基或任選地經烷基或滷素原子取代的雜芳基,
Y表示(CH2)n基團或O原子或S原子,且其中n=0-3,
且
R4表示式a.)的基團
項3.通式(V)的化合物,
-其中在式中
用虛線標示的鍵可以是單鍵或雙鍵,在位置5,6及13,14為雙鍵的情形下,其可以是順式或反式取向,
Q表示羥基且Z表示羥基或氧代基團,
R3表示直鏈或支鏈、飽和或不飽和的C4-6烴基,或C4-10烷基環烷基或環烷基,或任選地經烷基或滷素原子取代的苯基、任選地經烷基或滷素原子取代的C7-10烷基芳基或任選地經烷基或滷素原子取代的雜芳基,
Y表示(CH2)n基團或O原子或S原子,且n=0-3,
且
R5表示式b.)、c.)、d.)或e.)的基團,
其中(X)表示滷素原子或氫原子。
項4.項1的方法i)的方法,其特徵在於合適引入該基團R4的化合物是1,1』-羰基二咪唑或1,1』-硫羰基二咪唑。
項5.項1的方法ii)的方法,其特徵在於對於在存在活性劑的給定情形下引入基團R5,使用N-羥基琥珀醯亞胺、N-羥基鄰苯二甲醯亞胺、N-羥基-5-降冰片烯-內-2,3-二甲醯胺、1-羥基苯並三唑、(苯並三唑-1-基氧基)三(二甲基氨基)鏻六氟磷酸鹽、N,N』-二琥珀醯亞胺基碳酸酯或N,N』-二琥珀醯亞胺基草酸酯。
項6.項5的方法,其特徵在於所施用的活化劑是N,N』-二異丙基碳二亞胺、N,N』-二環己基碳二亞胺或2-氯-1,3-二甲基咪唑啉鎓氯化物。
項7.項1的方法,其特徵在於該反應是在醚型、芳族或極性非質子溶劑或它們的混合物中進行。
項8.項1的方法,其特徵在於引入基團R4是在20至80℃之間的溫度進行,優選在70℃的溫度進行。
項9.項1的方法i)的方法,其特徵在於式(III)與式(IV)化合物的反應是在20至80℃之間的溫度進行。
項10.項1的方法ii)的方法,其特徵在於該反應是在醚型、芳族或極性非質子溶劑或它們的混合物中進行。
項11.項1的方法ii)的方法,其特徵在於引入基團R5是在0至80℃之間的溫度進行。
項12.項1的方法ii)的方法,其特徵在於式(V)與式(IV)化合物的反應是在20至80℃之間的溫度進行,優選在室溫進行。
項13.項1的方法i)或ii)的方法,其特徵在於式(III)或式(V)的活化的羧酸衍生物通過結晶和/或色譜法純化。
項14.項1的方法iii)的方法,其特徵在於該反應是在醚型、芳族或極性非質子溶劑或它們的混合物中進行。
項15.項7、10或14的方法,其特徵在於使用四氫呋喃作為溶劑。
項16.項1的方法iii)的方法,其特徵在於該反應是在0至70℃之間的溫度進行。
項17.項1的方法iii)的方法,其特徵在於使用的鹼是N-甲基嗎啉、N,N-二異丙基乙基胺、1,5-二氮雜二環[4,3,0]壬-5-烯或1,8-二氮雜二環[5,4,0]十一碳-7-烯。
項18.項1的方法,其特徵在於所使用的式(IV)的鹼是乙胺。
項19.項1的方法i)、ii)或iii)的方法,其特徵在於該反應是在一鍋法條件下進行。
項20.用於製備通式(IIA)化合物的方法,
其中在式中
R3表示直鏈或支鏈、飽和或不飽和的C4-6烴基,或C4-10烷基環烷基或環烷基,或任選地經烷基或滷素原子取代的苯基、任選地經烷基或滷素原子取代的C7-10烷基芳基或任選地經烷基或滷素原子取代的雜芳基,
Y表示(CH2)n基團或O原子或S原子,且n=0-3,
所述方法的特徵是將通式(XII)的內酯二醇:
其中R3、Y及n的含義如上述定義,
還原成通式(XIII)的內酯三醇:
其中R3、Y及n的含義如上述定義,
移除通式(XIII)化合物的保護基,並將所得的通式(XIV)的化合物:
其中R3、Y及n的含義如上述定義,
通過Wittig反應而轉化成通式(IIA)的化合物。
項21.通式(XIII)的內酯三醇,
其中R3表示直鏈或支鏈、飽和或不飽和的C4-6烴基,或C4-10烷基環烷基或環烷基,或任選地經烷基或滷素原子取代的苯基、任選地經烷基或滷素原子取代的C7-10烷基芳基或任選地經烷基或滷素原子取代的雜芳基,
Y表示(CH2)n基團或O原子或S原子,且n=0-3。
項22.結晶形式的式(IIB)化合物,
項23.用於製備結晶形式的式(IIB)化合物的方法,其特徵在於向含有式(IIB)化合物的混合物中加入酯型與醚型溶劑的混合物。
項24.項23的方法,其特徵在於使用甲醚、乙醚、二異丙醚優選乙醚及二異丙醚作為醚型溶劑。
項25.項23的方法,其特徵在於使用乙酸乙酯、乙酸甲酯、乙酸異丙酯優選乙酸異丙酯作為酯型溶劑。
項26.項23的方法,其特徵在於結晶是在(-)30℃及30℃之間的溫度進行,優選在0至25℃之間的溫度進行。
項27.項23的方法,其特徵在於將晶體的混懸液攪拌1至24小時,優選8小時。
項28.項23的方法,其特徵在於將晶體混懸液過濾並用醚型溶劑洗滌,優選用二異丙醚洗滌。
項29.項23的方法,其特徵在於將該過濾的晶體在25至50℃之間真空乾燥,優選在35至40℃之間真空乾燥。
項30.用於製備式(IB)的比馬前列素的結晶形式II的方法,
其特徵在於:在後處理及蒸發後,於含有比馬前列素的反應混合物或比馬前列素的任何結晶或非結晶形式或它們的任何比例的混合物中,加入計算量的醚型或質子溶劑,如果需要時將其暴露至機械效應,且隨後乾燥並均勻化。
項31.經項30的方法製備的結晶形式II,其特徵在於其熔點是在72至78℃之間。
項32.經項30的方法製備的結晶形式II,其特徵在於根據DSC研究,吸熱峰在72至79℃之間且其熔化熱高於75焦耳/克。
項33.項30的方法,其特徵在於使用20-60質量%的質子溶劑,優選是35質量%的質子溶劑。
項34.項30的方法,其特徵在於使用醇和/或水作為質子溶劑。
項35.項34的方法,其特徵在於使用甲醇或乙醇作為醇。
項36.項34的方法,其特徵在於使用水作為質子溶劑。
項37.項30的方法,其特徵在於使用2000-8000質量%的醚型溶劑。
項38.項30方法,其特徵在於使用二甲醚、乙醚、二異丙醚作為醚型溶劑。
項39.項30的方法,其特徵在於使用攪拌或刮擦或兩者作為機械效應
項40.項30的方法,其特徵在於添加的溶劑通過乾燥除去。
項41.項33及40的方法,其特徵在於乾燥在(-)60℃及70℃之間的溫度在真空下進行。
項42.項37及40的方法,其特徵在於乾燥在低溫優選在0至(-)50℃之間通過使得氮氣經過而進行。
產物的鑑定是藉助於下面的分析儀器而進行:
NMR光譜通過Bruker-Avance III-500MHz儀器而記錄,DSC曲線通過Mettler-Toledo DSC 1/700儀器而記錄,IR光譜通過Perkin-Elmer Spektrum400FT-IR光譜儀而記錄,MS光譜通過Shimadzu LC-MS-IT-TOF儀器而記錄。熔點通過Büchi Melting Point B-545裝置確定。
本發明的其它細節是在實施例中描述,但本發明不受限於這些實施例。
附圖說明
圖1.實施例1a的內酯三醇的IR光譜。
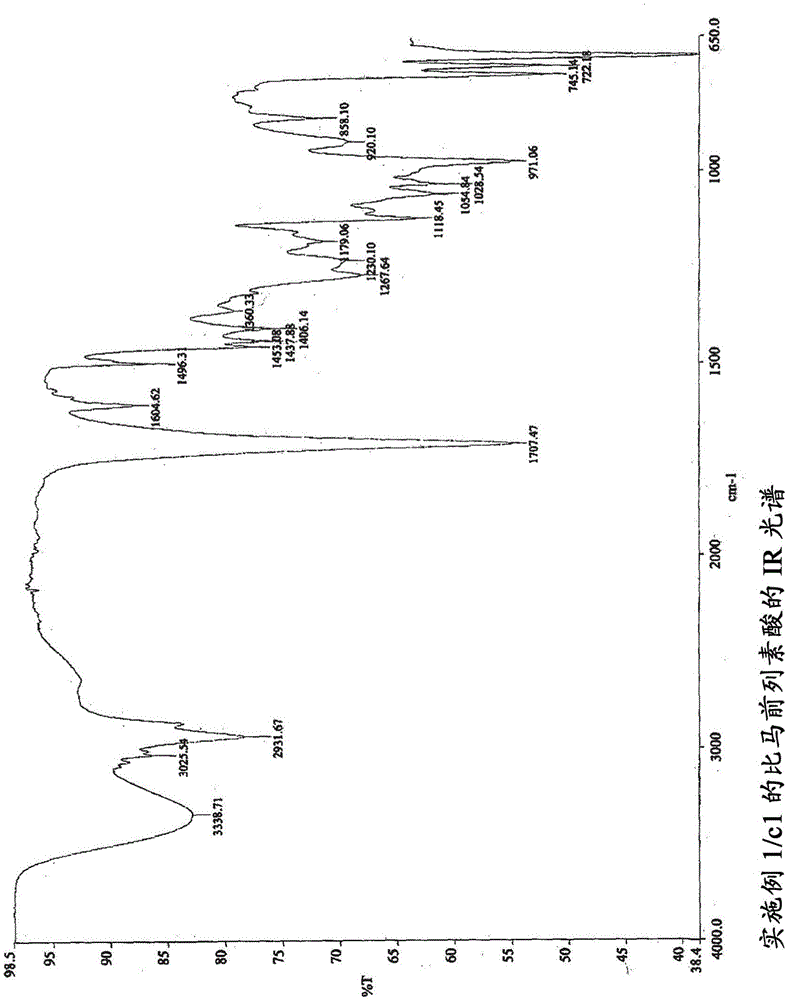
圖2.實施例1/c1的比馬前列素酸的IR光譜。
圖3.實施例14/a的形式I的IR光譜。
圖4.實施例10.)的形式II的IR光譜。
圖5.實施例14/a的形式I的DSC曲線。
圖6.實施例10的形式II的DSC曲線。
圖7.實施例14/a的形式I的X-射線衍射曲線。
圖8.實施例10的形式II的X-射線衍射曲線。
圖9.實施例1/c2.)的比馬前列素酸的X-射線衍射曲線。
圖10.實施例1/c2.)的比馬前列素酸的DSC曲線。
具體實施方式
1.製備起始物質
a.)製備[1,1』-聯苯]-4-羧酸((3aR,4R,5R,6aS)-六氫-4-[(1E,3S)-3-羥基-5-苯基-1-戊烯-1-基]-2-羥基-2H-環戊二烯並[b]呋喃-5-基)酯(PPB-內酯三醇)
將55克的[1,1』-聯苯]-4-羧酸((3aR,4R,5R,6aS)-六氫-4-[(1E,3S)-3-羥基-5-苯基-1-戊烯-1-基]-2-氧代-2H-環戊二烯並[b]呋喃-5-基)酯(PPB-內酯二醇)的內酯基
在1000毫升四氫呋喃(THF)溶劑中在(-)65-(-)85℃用422毫升二異丁基氫化鋁的己烷溶液(DIBAL-H)還原。將反應混合物用NaHSO4溶液破壞,將水相用乙酸乙酯萃取,將有機相用NaHCO3溶液洗滌並在40-50℃將溶劑除去。將粗物質蒸發後得到46.2克油狀物。
所得的PPB-內酯三醇的結構式:
在叔丁基甲基醚(TBME):己烷混合物中結晶後,從粗油狀物得到41.6克白色結晶。
熔點:91.1-91.7℃
實施例1a的內酯三醇的IR光譜如圖I所示。
13C及1H NMR數據:
*重疊的1H NMR信號(在括號中的數字是指信號基團在PMR光譜中的位置編號,方向:朝向減少的位移)
**13C NMR信號與DMSO溶劑的多重譜線重疊。
b.)製備(3aR,4R,5R,6aS)-六氫-4-[(1E,3S)-3-羥基-5-苯基-1-戊烯-1-基]-2H-環戊二烯並[b]呋喃-2,5-二醇(內酯三醇)
將46.2克的[1,1』-聯苯]-4-羧酸((3aR,4R,5R,6aS)-六氫-4-[(1E,3S)-3-羥基-5-苯基-1-戊烯-1-基]-2-羥基-2H-環戊二烯並[b]呋喃-5-基)酯(PPB-內酯三醇)油狀物溶解在230毫升甲醇中且在加入6.6克K2CO3後在35-45℃去醯基化。在(-)5-0℃用0.5M磷酸溶液將反應混合物的pH調整至7-8。將沉澱的結晶濾出並用甲醇:水混合物洗滌。將母液蒸發,用乙酸乙酯萃取,將有機相通過Na2SO4乾燥,將乾燥的物質濾出並將產物通過加入己烷而結晶。得到26克白色結晶的物質。
產物的結構式:
熔點:98-103℃
13C及1H NMR數據:
*重疊的1H NMR信號(在括號中的數字是指信號基團在PMR光譜中的位置編號,方向:朝向減少的位移)。
c.)製備7-[(1R,2R,3R,5S)-3,5-二羥基-2-[(1E,3S)-3-羥基-5-苯基-1-戊烯-1-基]-環戊基]-5-庚烯酸,(5Z)-(比馬前列素酸):
c1.)將108克4-羧基丁基溴化鏻(KBFBr)溶解在800毫升THF中並將溶液冷卻至0-(-)5℃。在此溶液中先加入91克叔丁醇鉀(KOtBu),且攪拌並冷卻至(-10)-(-15)℃後,加入25克內酯三醇在THF中的溶液。當達到預期的轉化後,用水將反應混合物破壞,隨後加入EtOAc。將水相用EtOAc洗滌。將水相用NaHSO4溶液酸化至pH=2並用EtOAc萃取。將合併的有機相用15%NaCl溶液洗滌,通過Na2SO4乾燥,過濾並蒸發。將殘留物從乙酸乙酯與二異丙醚的混合物結晶。將結晶濾出並洗滌,將濾液蒸發。將所得的黃色油狀物在矽膠上通過色譜法(用二異丙醚-丙酮洗脫劑)純化。得到25.5克油狀物。
所得的比馬前列素酸的IR光譜如圖II所示。
c2.)將實施例1/c1.)所得的產物溶解在60毫升乙酸異丙酯中並在攪拌下在其中加入40毫升乙醚。將少量的比馬前列素酸的晶種添加至反應混合物中。在攪拌下逐漸冷卻至0℃並在其中加入約60毫升二異丙醚。將混懸液在此溫度攪拌一夜後過濾並用二異丙醚洗滌且在真空乾燥,得到20.4克晶狀比馬前列素酸。
所得的比馬前列素酸的DSC曲線顯示在圖X且X-射線粉末衍射曲線是在圖IX。
產物的結構式:
熔點:63.0-65.5℃
13C及1H NMR數據:
*部分或完全重疊的1H NMR信號(在括號中的數字是指信號基團在PMR光譜中的位置編號,方向:朝向減少的位移)。
2.)製備7-[3,5-二羥基-2-(3-羥基-5-苯基-戊-1-烯基)-環戊基]-5-庚烯酸(2,5-二氧代-吡咯烷-1-基)酯(活化的酯)
將27.5克實施例1/c2.)的比馬前列素酸溶解在270毫升THF中並在室溫在其中加入13.7克N,N』-二異丙基碳二亞胺,隨後加入13.7克N-羥基琥珀醯亞胺。將混合物在此溫度攪拌後倒在1N NaHSO4溶液及叔丁基甲基醚(TBME)的混合物上。分離各相。將有機相用1N NaHCO3溶液洗滌,將水性-鹼性相用TBME萃取。將合併的有機相通過Na2SO4乾燥,過濾並蒸發。將殘留物從己烷:丙酮的混合物結晶,得到30.04克白色結晶狀物質。
產物:
熔點:93.5-103.4℃
3.)將27.5克實施例1/c1.)的比馬前列素酸溶解在270毫升THF中並在室溫在其中加入11.5克碳酸鉀及19.6克N,N』-二琥珀醯亞胺基碳酸酯。將反應混合物在攪拌下逐漸加熱至60℃且隨後倒在1N NaHSO4溶液及叔丁基甲基醚(TBME)的混合物上。分離各相。將有機相用1N NaHCO3溶液洗滌,將水性-鹼性相用TBME萃取。將合併的有機相通過Na2SO4乾燥,過濾並蒸發。將殘留物從己烷:丙酮的混合物結晶,得到30.9克白色結晶狀物質。
產物:
熔點:93.5-103.4℃
13C及1H NMR數據:
*重疊的13C NMR,DMSO信號。**,***,+,++,+++,#:重疊的1H NMR信號。
4.)製備7-[3,5-二羥基-2-(3-羥基-5-苯基-戊-1-烯基)-環戊基]-5-庚烯酸1,3-二氧代-1,3-二氫-異吲哚-2-基酯(活化的酯)
將2克的比馬前列素酸溶解在20毫升THF中並在室溫在此溶液中加入1克N-羥基-鄰苯二甲醯亞胺和1毫升N,N』-二異丙基碳二亞胺。將反應混合物攪拌2小時後倒在1N NaHSO4溶液及叔丁基甲基醚(TBME)的混合物上。分離各相。將有機相用1N NaHCO3溶液洗滌,將水性-鹼性相用TBME萃取。將合併的有機相通過Na2SO4乾燥,過濾並蒸發。將殘留物從己烷:丙酮的混合物結晶,得到1.5克白色結晶狀物質。
產物:
熔點:83.2-84.5℃
13C及1H NMR數據:
$DMSO信號重疊的13C NMR。$$重疊的13C NMR信號。*,**,***,+,++:重疊的1H NMR信號。
5.)通過活化的酯製備7-[(1R,2R,3R,5S)-3,5-二羥基-2-[(1E,3S)-3-羥基-5-苯基-1-戊烯基]-環戊基]-N-乙基-5-庚烯醯胺,(5Z(-)比馬前列素)
將27.5克的比馬前列素酸溶解在270毫升THF中並在室溫在此溶液中加入13.7克N,N』-二異丙基碳二亞胺,隨後加入13.7克N-羥基琥珀醯亞胺。將混合物在室溫攪拌。所得的活化酯不分離。
完成酯形成後,將70毫升乙胺在THF中的2M溶液添加至反應混合物中。將混合物攪拌直到達成預期的轉化,然後將其倒在1N NaHSO4溶液及叔丁基甲基醚(TBME)的混合物上。分離各相。將有機相用1N NaHCO3溶液洗滌,將水性-鹼性相用TBME萃取。將合併的有機相通過Na2SO4乾燥,過濾並蒸發後得到25.4克油狀物。
產物:
13C及1H NMR數據:
MS數據
MS光譜:
正離子化:
預期的化學式:
C25H37NO4
測量的實際質量:438.2648[M+Na]+
預期的實際質量:438.2615[M+Na]+ΔM=3.3mDa及7.53ppm
C25H35NO3(M-H2O)
測量的實際質量:398.2655[M-H2O+H]+
預期的實際質量:398.2690[M-H2O+H]+ΔM=-3.5mDa及8.79ppm
MSMS(先驅離子:438.26)
預期的化學式:
C25H35NO3(M-H2O)
測量的實際質量:420.2520[M-H2O+Na]+
預期的實際質量:420.2509[M-H2O+Na]+ΔM=1.1mDa及2.62ppm
C25H32NO3(M-H2O-5H)
測量的實際質量:394.2366[M-H2O-5H]+
預期的實際質量:394.2377[M-H2O-5H]+ΔM=-1.1mDa及2.79ppm
C25H30NO2(M-2xH2O-5H)
測量的實際質量:376.2258[M-2xH2O-5H]+
預期的實際質量:376.2271[M-2xH2O-5H]+ΔM=-1.3mDa及3.46ppm
6.)通過活化的酯製備比馬前列素
將27.5克的比馬前列素酸溶解在270毫升THF中並在室溫在此溶液中加入11.5克碳酸鉀及19.6克N,N』-二琥珀醯亞胺基碳酸酯。將反應混合物在攪拌下逐漸加熱至60℃。所得的活化酯不分離。
活化的酯形成後,將70毫升乙胺在THF中的2M溶液添加至反應混合物中。當反應完成後將混合物倒在1N NaHSO4溶液及EtOAc的混合物上。將有機相用1N NaHCO3溶液洗滌,將水性-鹼性相用EtOAc萃取。將合併的有機相用NaCl溶液洗滌並通過Na2SO4乾燥。將乾燥的物質濾出,將濾液蒸發後得到25.7克油狀物。
產物:
7.)通過活化的醯胺製備比馬前列素
將27.5克的比馬前列素酸溶解在270毫升吡啶中並在其中加入13.7克1,1』-羰基二咪唑。將混合物在20-25℃攪拌直到發生活化的醯胺形成。所得的活化醯胺不分離。
將70毫升乙胺在THF中的2M溶液添加至在室溫的反應混合物中並將混合物攪拌直到達成預期的轉化。然後將混合物倒在1N NaHSO4溶液及叔丁基甲基醚(TBME)的混合物上。分離各相,將有機相用1N NaHCO3溶液洗滌並將水性-鹼性相用TBME萃取。將合併的有機相通過Na2SO4乾燥,過濾並將濾液蒸發後得到23.82克油狀物。
產物:
8.)從純化的經活化的酯製備比馬前列素
將30.9克實施例3的經活化的酯溶解在270毫升THF中並在此溶液中加入70毫升乙胺溶解在THF中的2M溶液。反應完成後,將混合物倒在1N NaHSO4溶液及EtOAc的混合物上。將有機相用1N NaHCO3溶液洗滌。將水性-鹼性相用EtOAc萃取。將合併的有機相用NaCl溶液洗滌並通過Na2SO4乾燥。將乾燥的物質濾出並將濾液蒸發。在所得的油狀物中加入35質量%的水且將產物結晶。得到24.8克高於99.5%純度的白色比馬前列素結晶。
產物:
熔點:71.9-72.5℃
HPLC:99.6%比馬前列素,低於0.3%反式-比馬前列素,0.1%其它雜質
9.)根據方法iii.)製備比馬前列素
將2.00克比馬前列素酸溶解在20毫升四氫呋喃(THF)中並在30℃先加入1.29克2-氯-1,3-二甲基咪唑啉鎓氯化物及1.44毫升三乙胺,經10分鐘攪拌後,加入2.57毫升乙胺在THF中的2M溶液。將反應混合物在1小時內逐漸加熱至70℃並將混合物在此溫度攪拌直到起始物質消失(約1小時)。通過TLC追蹤反應。
反應完成後,將混合物倒在1N NaHSO4溶液及乙酸異丙酯(iPrOAc)的混合物上。將有機相用1N NaHCO3溶液洗滌,將水性-鹼性相用iPrOAc萃取。將合併的有機相用NaCl溶液洗滌並通過Na2SO4乾燥。將乾燥的物質濾出並將濾液蒸發後得到1.41克油狀物
產物:
10.)從粗比馬前列素油狀物製備比馬前列素的結晶形式II
在根據實施例6製備的比馬前列素油狀物中,加入35質量%的純水。將混合物充分攪拌後在最高35℃的溫度真空乾燥,且每2小時攪拌並刮擦。完成乾燥後,將混合物均勻化。此產物的IR光譜顯示在圖IV且此產物的DSC曲線顯示在圖VI。生產的形式II的X-射線衍射曲線顯示在圖VIII。
收率:96.9%
熔點:78℃
DSC起點:73.56℃
11.)製備比馬前列素的結晶形式II
將根據實施例5製備的粗比馬前列素在加熱下溶解在3000倍量的乙醚中。然後在(-)20-(-)30℃通過使得氮氣緩慢通過將溶劑除去。將所得的結晶均勻化,或先暴露至機械效應且隨後均勻化。
收率:94.4%
熔點:75.9℃
DSC起點:72.92℃
12.)製備比馬前列素的結晶形式II
在根據實施例6製備的粗比馬前列素中加入35質量%的甲醇。將混合物充分攪拌後在最高35℃的溫度真空乾燥,且每2小時攪拌並刮擦。完成乾燥後,將混合物均勻化。
收率:95.8%
熔點:77.2℃
DSC起點:73.07℃
13.)製備比馬前列素的結晶形式II
在根據實施例6製備的粗比馬前列素中加入17.5質量%的純水及17.5質量%的乙醇。將混合物充分攪拌後在最高35℃的溫度真空乾燥,且每2小時攪拌並刮擦。完成乾燥後,將混合物均勻化。
收率:92.3%
熔點:72.9℃
DSC起點:72.96℃
14.)
a.)製備比馬前列素的結晶形式I(根據專利申請US 20090163596的實施例38)
將5.2克的粗比馬前列素從106克乙腈結晶:將混合物加熱至接近沸點的溫度,使熱溶液冷卻至室溫並將混合物在此溫度攪拌1小時,然後在0-5℃攪拌2小時。將沉澱的結晶濾出,用20克冷(0-5℃)乙腈洗滌並在0-5℃真空乾燥1小時,在室溫經半小時且在30-40℃經2小時。
得到4.3克的比馬前列素的結晶形式I。其IR光譜顯示在圖III,其DSC曲線顯示在圖V且其形式I的X-射線衍射曲線顯示在圖VII。
收率:83%
熔點:62.1℃
DSC起點:63.61℃
b.)從形式I開始製備比馬前列素的結晶形式II
在根據實施例14a製備的比馬前列素的結晶形式I中加入35質量%的純水。將混合物充分攪拌後在最高35℃的溫度真空乾燥,且每2小時攪拌並刮擦。完成乾燥後,將混合物均勻化。
收率:97.3%
熔點:77.7℃
DSC起點:73.14℃
15.)製備比馬前列素結晶形式II及I的混合物的結晶形式II
在結晶形式II及I的50%-50%混合物中,加入35質量%的純水。將混合物充分攪拌後在最高35℃的溫度真空乾燥,且每2小時攪拌並刮擦。完成乾燥後,將混合物均勻化。
收率:97.6%
熔點:78.2℃
DSC起點:73.77℃。