一種16‑羥基‑9‑烯‑十六羧酸酯的合成方法與流程
2023-08-13 03:45:01 1
本發明涉及有機化工領域,特別涉及一種16-羥基-9-烯-十六羧酸酯的合成方法。
背景技術:
對於自然界存在的香料化合物,其中最重要的一個類別就是具有烯烴結構的大環內類烯烴化合物。該類型化合物均具有優雅柔和的香型,定香性能良好,接近天然香料的典型氣味。由於直接從天然獲取該類型的化合物,成本比較高,並且對自然環境造成巨大損害。因此研究其簡單高效的合成方法成為發展的必然。由於該類型的化合物主要結構為大環酮類和內酯類化合物烯烴化合物,其合成的關鍵在於烯烴的生成與環化。對於烯烴化反應,經常採用鄰二醇的化合物經過脫水烯化而來,而對於分子內的成環化反應,根據原料結構的不同,其成環方式也有所區別,從成環的方式而言,主要包括了以醯氧鍵碳氧鍵及碳碳鍵等成環方式。對於酮式大環類化合物,通常可採用羥醛縮合法來合成製備,反應條件溫和,目前該類化合物的生產已得到較好解決。
而大環內酯烯化合物仍存在較大的合成難度,其已經作為合成大環麝香香料的研究熱點之一。大環內酯的基本結構是12元環以上且環骨架上含有一個或多個酯基的分子化合物,傳統的合成路線常使用滷代長鏈烷酸作為原料進行生產,但原料加工路線長,滷化物的添加導致環境汙染嚴重(2000,TL,41(45):8673)。
該方法先用15%的溴化氫乙酸溶液溴化,酯化,烯烴化,脫保護得到關鍵中間烯基羧酸類化合物,該化合物進一步環合或者即可製備酯化或酮基化產物。其溴化氫的過量使用,對生成設備和環境造成了極大的汙染,因此,使得人們逐漸將注意力轉移到綠色環保的合成方法研究的研究上來。
由於合成該類型化合物多採用多羥基脂肪羧酸作為起始原料,其分子中含有一個游離的羧基和三個游離的羥基,其本身能夠作為合成大環內酯的一種原料,在很大程度上減少了繁瑣冗餘有毒有害的中間合成過程,從而降低合成成本。但由於分子結構中端位的碳原子所連接的羧基及羥基基團,導致其成環的過程是以形成醯氧鍵的方式進行的,即活化分子中一端的羧基,使之與另一端的羥基反應生成大環內酯的方法。其直接的內酯化過程,除了存在分子間酯化連接外,其分子內產物亦存在如下三種可能的結構,其結構具有有一定的相似性,分離困難。
為了避免生成上述混合物,人們往往採用分步的方法進行合成,其已有合成方法主要可以分為兩種類型,第一種,主要採用選擇性的保護基,現對領位二羥基進行保護,然後端基環取代環化,再進行烯烴化獲取目標物質;第二種,類似於滷代合成類似的方法,先選擇合適的保護基團對碳鏈端頭的羥基和羧基進行保護,烯化,脫保護和環化得到目標產物。
第一種方法,常採用鄰二醇的縮醛保護,避免9和10位的內酯化的基礎上,再經過脫叉烯化,內酯化得到目標品,或者先內酯化,然後脫叉烯化反應得到目標產物。Shiina等在樟腦磺酸(CSA)的催化下(T,2006,62(33):7934;TL,2002,43(42):7535;TL,2010,51(17):2348;TL,1999,40(6):1123),紫膠酮酸與PhCH(OMe)2發生縮醛反應得苄叉保護中間產物,經活性酯——2-甲基-6-硝基苯甲酸酐(MNBA)和4-二甲氨基吡啶氧化物(DMAPO)催化發生內酯化反應獲得單體內酯中間產物,在醋酸催化下水解叉苄叉,其所得鄰二醇與硫代羰基二咪唑反應形成硫代碳酸酯,然後還原脫硫形成卡賓,分子內電子轉移脫掉一分子二氧化碳形成雙鍵的目標產物,其反應過程如下;在這個過程亦有研究者直接利用N,N-二甲基甲醯胺二新戊基乙縮醛(DMF-DEA)進行叉化保護。然後在酸酐的作用下,直接脫叉烯烴化得到目標產物,減少了反應步驟。
另外一種是利用具有大位阻效應的活性酯催化劑,選擇性的端基內酯化,然後再脫鄰二醇成烯烴化反應,如Venkataraman等(TL,1980,21(19):1893;TL,1979,20(32):3037)利用三聚氯氰(TCT)催化長鏈-羥基酸進行內酯化反應,在丙酮中先後加入三聚氯氰(TCT)和三胺(Et3N),混合於室溫條件下反應得到產物,如合成(9E)-異黃葵內酯的中間產物。其最終產物收率高達86%。同理由於N,N-二甲基甲醯胺二新戊基乙縮醛(DMF-DEA)對高位阻化合物能夠顯示出獨特的優越性,且能與很多含有活潑氫的化合物反應,已經越來越被人們用於醚硫醚等雜環化合物的合成中。Villemin在氮氣保護下(Synthesis,1987(2):154;TL,2006,47(31):5553),就利用DMF-DEA在甲苯混合,發生親電取代生成,然後經醋酸酐脫水減壓蒸餾收集得到目標產物。
以前所有製備方法均需要分步進行,反應步驟較長,生產成本高;且中間產物還需經過矽膠柱層析分離純化才可以得到適合下一步反應的原料,不利於大規模生產使用。
技術實現要素:
本發明的發明目的是提供一種16-羥基-9-烯-十六羧酸酯的合成方法。該方法中多羥基脂肪羧酸經過一鍋法進行端羥基和羧酸的保護,以及領羥基的烯烴活化,不經任何分離操作,加入催化劑直接將領羥基脫水烯烴化,反應結束直接加水分解保護基,即得到目標產物。且該方法操作簡單,反應所涉及到試劑對環境友好,目標產物分離純化簡單。
為了實現以上目的,本發明的技術方案為:
一種16-羥基-9-烯-十六羧酸酯的合成方法,該方法包括以下步驟:
(1)、採用多羥基脂肪酸作為反應起始原料,如9,10,16-三羥基十六羧酸(1.0eq);
(2)、選擇的保護試劑為原酸縮酸酯類化合物,如原甲酸三甲酯,原甲酸三乙酯或乙酸三甲酯等,其與原料的配比為2.5~10eq;
(3)、合成反應溫度為50~120℃,反應時間為2~8h;
(4)、烯烴化反應催化劑為各種類型的酸酐類催化劑,如乙酸酐,丙酸酐,丁酸酐和苯甲酸酐,其與原料的配比為1.0~2.0eq;
烯烴化反應溫度為110~180℃,反應時間為3~8h。
該方法具體包括以下步驟:
取1當量(1.0eq)9,10,16-三羥基十六羧酸加入到單口反應瓶中,然後加入3倍體積的矽油和2.5~10eq的原酸縮酸酯(原甲酸三甲酯),氮氣保護下,控溫在50~120℃反應2~8h;然後加入約1.0~2.0eq的酸酐,控溫在110~180℃反應約3~8h。冷卻到室溫,加入一定量的H2O,攪拌水解約30min,然後調節pH到5.0,乙酸乙酯萃取,無水硫酸鈉乾燥,旋轉蒸乾得到粗品16-羥基-9-烯-十六羧酸酯。
精製的方法包括以下步驟:
上述產品溶解在一定量的水中,用氫氧化鈉調節pH到9.0~9.5,樣品溶解完全,用石油醚萃取三次,然後水層調節pH到5.0,乙酸乙酯萃取,合併萃取液,無水硫酸鈉乾燥,過濾,旋轉蒸乾。固體中加入約一定量的無水乙醇重結晶獲得目標產物,NMR分析結構正確。
本發明的有益效果是:
(一)、本研究根據端基羥基和羧基位阻比較小的特點,開發出了一鍋法合成的該類型化合物關鍵中間體——16-羥基-9-烯-十六羧酸酯的合成方法。
(二)、其多羥基脂肪羧酸經過一鍋法進行端羥基和羧酸的保護,以及領羥基的烯烴活化,不經任何分離操作,加入催化劑直接將領羥基脫水烯烴化,反應結束直接加水分解保護基,即得到目標產物。
(三)、該方法操作簡單,反應所涉及到試劑對環境友好,目標產物分離純化簡單。
(四)、分離純化簡單,有利於大規模工業化生產。
附圖說明
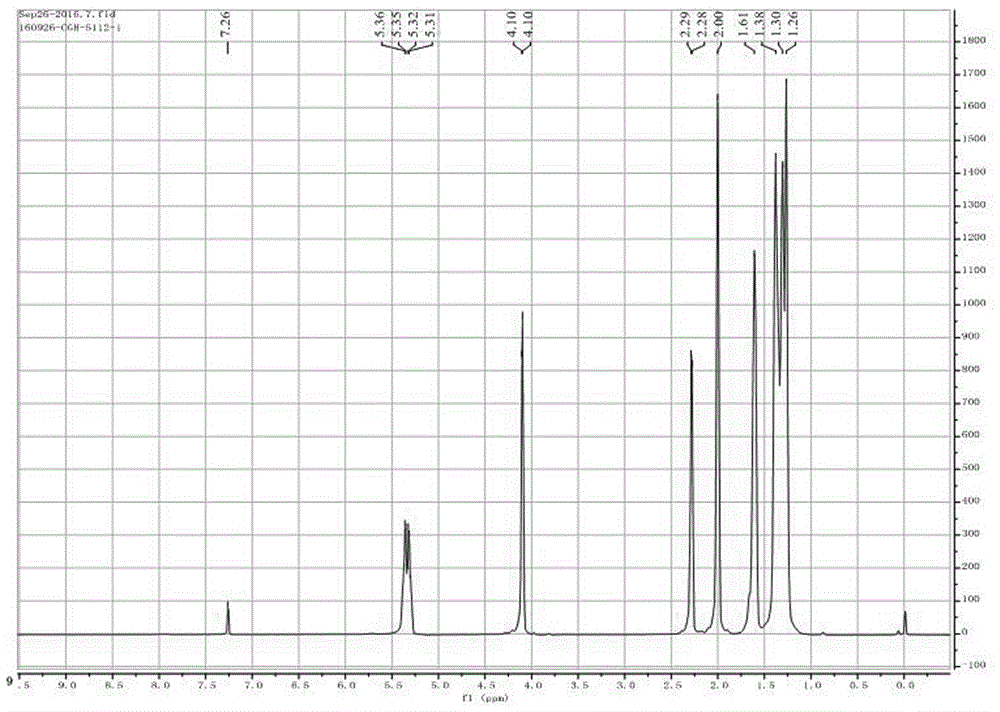
圖1為9-烯-十六羧酸內酯H1NMR圖
圖2為9-烯-十六羧酸內酯C13NMR圖
具體實施方式
下面結合具體實施例,進一步闡述本發明。應理解,這些實施例僅用於說明本發明而不用於限制本發明的範圍。此外應理解,在閱讀了本發明的內容之後,本領域技術人員可以對本發明作各種改動或修改,但這些等價形式同樣落於本申請所附權利要求書所限定的範圍。
實施例1:
取1.0g(3.3mmol)9,10,16-三羥基十六羧酸加入到單口反應瓶中,然後加入約3.0mL的矽油和1.0mL(3.0eq)的原甲酸三乙酯,氮氣保護下,控溫在50℃反應2h,然後升溫到120℃反應6h;冷卻低於50℃,迅速一次性的加入0.31mL(1.0eq)的乙酸酐,控溫在110℃反應h,然後升溫到180℃反應約7h。冷卻到室溫,加入30mL的H2O,攪拌水解約30min,然後調節pH到5.0,用乙酸乙酯萃取(20mL×3),無水硫酸鈉乾燥,旋轉蒸乾得到粗品16-羥基-9-烯-十六羧酸酯。
實施例2:
取1.0g(3.3mmol)9,10,16-三羥基十六羧酸加入到單口反應瓶中,然後加入約3.0mL的矽油和2.0mL(6.0eq)的原甲酸三乙酯,氮氣保護下,控溫在60℃反應4h,然後升溫到120℃反應6h;冷卻低於50℃,迅速一次性的加入0.47mL(1.5eq)的乙酸酐,控溫在110℃反應2h,然後升溫到180℃反應約7h。冷卻到室溫,加入30mL的H2O,攪拌水解約30min,然後調節pH到5.0,用乙酸乙酯萃取(20mL×3),無水硫酸鈉乾燥,旋轉蒸乾得到粗品16-羥基-9-烯-十六羧酸酯。
實施例3:
取1.0g(3.3mmol)9,10,16-三羥基十六羧酸加入到單口反應瓶中,然後加入約3.0mL的矽油和3.0mL(9.0eq)的原甲酸三乙酯,氮氣保護下,控溫在70℃反應3h,然後升溫到120℃反應6h;冷卻低於50℃,迅速一次性的加入0.56mL(1.8eq)的乙酸酐,控溫在110℃反應1h,然後升溫到180℃反應約7h。冷卻到室溫,加入30mL的H2O,攪拌水解約30min,然後調節pH到5.0,用乙酸乙酯萃取(20mL×3),無水硫酸鈉乾燥,旋轉蒸乾得到粗品16-羥基-9-烯-十六羧酸酯。
實施例4:
取1.0g(3.3mmol)9,10,16-三羥基十六羧酸加入到單口反應瓶中,然後加入約3.0mL的矽油和4.0mL(10.0eq)的原甲酸三乙酯,氮氣保護下,控溫在80℃反應2h,然後升溫120℃反應6h;冷卻低於50℃,迅速一次性的加入0.62mL(2.0eq)的乙酸酐,控溫在110℃反應1h,然後升溫到180℃反應約7h。冷卻到室溫,加入30mL的H2O,攪拌水解約30min,然後調節pH到5.0,用乙酸乙酯萃取(20mL×3),無水硫酸鈉乾燥,旋轉蒸乾得到粗品16-羥基-9-烯-十六羧酸酯。
精製:
取實施例1中獲取的0.6g產品16-羥基-9-烯-十六羧酸酯溶解在約30mL的水中,用5wt%的氫氧化鈉溶液調節pH到9.0~9.5,樣品溶解完全,用石油醚萃取3次,每次20ml,即(20mL×3),然後水層用1.0M的鹽酸調節pH到5.0,乙酸乙酯萃取(20mL×3),合併萃取液,無水硫酸鈉乾燥,過濾,旋轉蒸乾。固體中加入約15mL的無水乙醇重結晶獲得目標產物約0.67g,收率為75%。
驗證案例:
上述精製的16-羥基-9-烯-十六羧酸酯約1.0g加入到單口瓶中,加入20mL的無水甲苯和0.2g的對甲苯磺酸,加熱回流分水反應約4h,冷卻到室溫,加入約20mL的H2O洗滌,調節水層pH到中性(20mL×2),無水硫酸鈉乾燥,過濾,旋轉蒸乾得到無色油狀液體,NMR分析目標產物9-烯十六環內酯(13C NMR(151MHz,Chloroform-d)δ174.20,131.33,130.75,64.52,35.06,31.94,31.57,29.79,29.05,28.73,28.14,28.09,28.02,27.10,26.94,25.19.1H NMR(600MHz,Chloroform-d)δ5.34(dd,J=21.9,6.8Hz,2H),4.10(d,J=4.5Hz,2H),2.28(d,J=7.0Hz,3H),2.00(s,4H),1.61(s,3H),1.49–1.12(m,14H))。
以上所述實例僅是本專利的優選實施方式,但本專利的保護範圍並不局限於此。應當指出,對於本技術領域的普通技術人員來說,在不脫離本專利原理的前提下,根據本專利的技術方案及其專利構思,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本專利的保護範圍。