一種6a‑甲基氫化可的松的製備方法與流程
2023-04-30 19:13:53 1
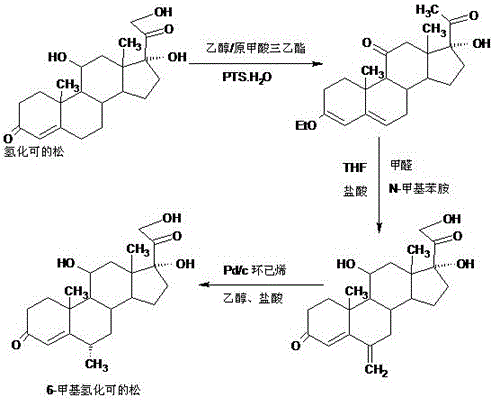
本發明屬於甾體激素藥物及其中間體的製備工藝技術,具體涉及一種甲基潑尼松龍的關鍵中間體6a-甲基氫化可的松的製備方法。
背景技術:
甲基潑尼松龍(分子式C22H32O5),化學名為6a-甲基-11b,17a,21-三羥基-孕甾-1,4-二烯-3,20-二酮,是一種第二代甾體糖皮質激素藥物。在臨床上主要用於原發及繼發性腎上皮質功能減退、風溼病、天胞瘡、急性支氣管哮喘、過敏性鼻炎、紅斑狼瘡、休克、兒童急性白血病、腫瘤引起的高鈣血症等許多疾病的治療,副作用低,效果好,市場前景廣闊。6a-甲基氫化可的松是生產甲基潑尼松龍的關鍵中間體,將其以DDQ1位脫氫或其他方法1位脫氫即得到甲基潑尼松龍。
6a-甲基潑尼松龍的傳統生產方法是,以薯蕷植物中提取的薯蕷皂素,經六步化學反應,一步微生物發酵,再經三步化學反應獲得的重要激素藥物中間體21去氧可的松為原料,經3位烯醚化、6位次甲基化、6位氫化、3,20位縮酮保護、11位還原、脫保護等六步反應,製得關鍵中間體P6,P6經上碘、置換兩步反應,製得6a-甲基氫化可的松,其工藝路線見附圖1。其合成路線長,合成總收率低,而且其中薯蕷皂素的提取及其後多步合成化學反應產生廢水較多,且不易處理,易汙染環境。更重要的是,隨著野生薯蕷植物資源日益枯竭,而種植薯蕷植物因人工、化肥等種植成本的日益上升,導致皂素、黴氧的生產成本成倍增長,致使6-甲基氫化可的松及甲基潑尼松龍的生產成本成倍增長,業已對甲基潑尼松龍的全球市場產生了重大影響。
技術實現要素:
本發明的目的是提供以氫化可的松為原料,經三步反應合成6-甲基氫化可的松的製備方法,大幅提高合成總收率,顯著降低其生產成本,避免6a-甲基氫化可的松傳統生產工藝中生產原料貴、汙水處理難、工藝操作複雜,生產成本高等諸多缺陷。
本發明的技術方案是:6a-甲基氫化可的松的製備方法,採用由4-雄烯二酮(簡稱4AD)製備的氫化可的松為原料,經3位醚化,6位次甲基化,氫化,三步反應合成6a-甲基氫化可的松,即:
A、合成醚化物,用由4AD製備的氫化可的松,在有機溶劑中與原甲酸三乙酯酸催化反應,得醚化物:3-乙烯醚化氫化可的松;
B、合成次甲基物,將醚化物在有機溶劑中跟N-甲基苯胺與甲醛發生滿裡希反應,得次甲基物:6-次甲基氫化可的松;
C、合成6a-甲基氫化可的松,將次甲基物在有機溶劑中進行催化氫化反應,得6a-甲基氫化可的松。
進一步地,一種6a-甲基氫化可的松製備方法,具體操作步驟如下:
A、合成醚化物:是將氫化可的松溶解於有機溶劑中,於40-70℃下加入原甲酸三乙酯,強酸催化下反應6~8小時,TLC確認反應終點,反應完後,加鹼中和,回收有機溶劑,然後以乙醇水溶液結晶,乾燥得醚化物:3-乙醚基烯醇式氫化可的松,HPLC含量97.0-99.0%,重量收率100-105%;
B、合成次甲基物:是將上述醚化物溶入有機溶劑中,加入N-甲基苯胺和37%的甲醛水溶液,攪拌,再加入酸催化劑,保溫於25~30℃反應1-2小時,再保溫於50-70℃繼續反應1-1.5小時,TLC確認反應終點,反應完後,加入適量的鹼調pH7-8,減壓濃縮有機溶劑至幹,然後加入甲醇,攪拌,降溫至0-10℃,再慢慢滴加濃鹽酸,加完後保溫於20-30℃繼續反應16-18小時,TLC確認反應終點,反應完後,減壓濃縮約80%的甲醇,冷卻,加入相當於醚化物3-4倍的自來水,攪拌析晶2-3小時,離心,洗滌,洗液與濾液減壓回收有機溶劑後排入廢水處理池,濾餅70℃以下乾燥,得次甲基物粗品,HPLC含量96.0-97.5%,重量收率100-105%;粗品經C4以下低碳醇水溶液重結晶,得次甲基物精品,HPLC含量99.0%以上,精製重量收率80-85%,本步重量總收率82-85%;
C、合成6a-甲基氫化可的松: 是將上述次甲基物溶入有機溶劑中,加入鈀炭催化劑,氮氣置換後,保溫於45-65℃下,慢慢加入環己烯,0.5-1.0小時內加完,加完後再在45-65℃下保溫反應1.5-2小時,HPLC確認反應終點,反應完後,立即濾除鈀炭,降溫至25-30℃,慢慢滴加6N濃酸,0.5-1.0小時內滴完,然後再保溫於45-65℃繼續轉位反應1.5-2小時,HPLC確認反應終點,反應完後,加入10%燒鹼溶液中和至溶液的pH6.5-7.5,減壓濃縮回收90-95%的有機溶劑後,冷卻降溫至10-25℃,加入相當所投次甲基物物2倍的自來水,攪拌析晶1.5-2.5小時,離心,洗滌,洗液與濾液排入廢水處理池,濾餅100℃以下乾燥,得6a-甲基氫化可的松粗品,HPLC含量98.0-99.0%,重量收率80-85%;粗品經C4以下低碳醇重結晶,得6a-甲基氫化可的松精品,HPLC含量99.0%以上,精製重量收率85-90%,本步重量總收率70-75%;三步合成6a-甲基氫化可的松的總收率60-65%。
上述6a-甲基氫化可的松製備方法,進一步說明如下:
A、醚化物合成中所述有機溶劑,包括二氯甲烷、甲苯、C4以下低碳醇,酸催化劑包括鹽酸、硫酸、磷酸,或醋酸、對甲苯磺酸、草酸;中和所用弱鹼包括碳酸鈉或吡啶,反應溫度20~70℃;反應物的重量配比是,氫化可的松:原甲酸三乙酯:酸=1g:0.5~1.0g: 0.01~0.05g;反應物與溶劑間的配比是,氫化可的松:有機溶劑=1g:4~8ml;
B、次甲基物合成中所述有機溶劑包括:甲苯、二氯甲烷、氯仿、DMF、四氫呋喃、二氧六環、C4以下低碳醇或它們的混合物,滿裡希反應所用的酸包括鹽酸、硫酸、磷酸或醋酸、對甲苯磺酸、草酸;中和所用鹼包括吡啶或三乙胺,滿裡希反應的反應溫度10~40℃,滿裡希反應物的重量配比是,醚化物:N-甲基苯胺:甲醛:酸=1g:0.5~1.0ml:2.0~2.6ml: 0.01~0.05g;反應物與溶劑間的配比是,醚化物:有機溶劑=1g:5~15ml,水解反應溫度10-40℃,水解反應的各物料配比是醚化物:甲醇:酸=1g:5~10ml: 1.2~1.5g;
C、6a-甲基氫化可的松合成中所述有機溶劑包括:甲苯、二氯甲烷、四氫呋喃、乙酸乙酯、DMF、冰醋酸、C4以下低碳醇;氫化反應催化劑是鈀炭或活性鎳;反應溫度是45-65℃;轉位反應的酸包括硫酸、鹽酸,反應溫度為10-40℃;氫化反應物間的重量配比是,次甲基物:氫化試劑:催化劑=1g:1.5~2.5ml:0.1-0.3g,氫化反應反應物與溶劑間的配比是:次甲基物:有機溶劑=1g:3-5ml,水解反應所需酸的配比是:次甲基物:酸=1g:2-4ml。
該6a-甲基氫化可的松製備方法的優先方法是:
醚化物合成中所述有機溶劑是乙醇;酸催化劑用對甲苯磺酸;中和反應溫度40~50℃,反應物的重量配比是,氫化可的松:原甲酸三乙酯:酸=1g:0.8g:0.02g;
次甲基物合成中所述有機溶劑是二氯甲烷或乙醇;滿裡希反應所用的酸是對甲苯磺
酸,反應溫度25~30℃;滿裡希反應物的重量配比是,醚化物:N-甲基苯胺:甲醛:酸=1g:0.8ml:2.2ml:0.02g;反應物與溶劑間的配比是1g::8~10ml;水解反應溫度20-25℃;甲醇:酸=1g:8ml:1.25g;
6a-甲基氫化可的松合成中所述有機溶劑是乙醇,氫化反應溫度是55-60℃;轉位反應的酸是鹽酸,反應溫度是20-25℃;次甲基物:氫化試劑:催化劑=1g:2ml:0.2g;反應物與溶劑間的配比是,次甲基物:有機溶劑=1g:4ml;次甲基物:酸=1g:3ml。
本發明的有益效果是:以4AD製備的氫化可的松為原料,經3位醚化,6位次甲基化,氫化等三步反應合成6a-甲基氫化可的松,相對以薯蕷皂素加工獲得的黴脫物作原料的傳統方法,本發明工藝具有原料來源廣泛,工藝經濟環保,生產操作簡便,合成路線短,產品收率高等諸多優點;以本法生產6a-甲基氫化可的松,生產成本比傳統方法降低40-50%;生產中使用的溶劑可回收循環套用,易實施工業化生產。
附圖說明
圖1是傳統方法6a-甲基氫化可的松的合成路線圖;
圖2是本發明6a-甲基氫化可的松的合成路線圖。
具體實施方式
為了更方便地說明本發明的要點與精神,下面舉例予以說明:
實施例一:
A、醚化物的製備
在一個1000ml三口瓶中,加入100g 氫化可的松、200ml乙醇、80ml原甲酸三乙酯、2g對甲苯磺酸,保溫於40-45度攪拌反應6~8小時,TLC檢測反應終點,反應完後,加入3ml 吡啶,攪拌20-25分鐘中和酸,然後減壓回收90-95%的有機溶劑,再加入500ml 50%的乙醇水溶液,將體系冷卻到-5-0度,攪拌結晶2~3小時,抽濾,少量乙醇水溶液洗滌,洗液和濾液合併,回收溶劑,濾餅70度以下烘乾,得醚化物101.6g,HPLC含量99.2%,重量收率101.6%。
B、次甲基物的製備
在一個1000ml三口瓶中,加入100g 醚化物、800ml乙醇、50ml二氯甲烷,溶解後加入2g對甲苯磺酸與50ml乙醇的溶液 ,然後再加入70mlN-甲基苯胺與200ml 37%的甲醛溶液,升溫至25-30度保溫攪拌反應1.5~2.0小時,再在50-55度繼續反應1.0-1.5小時,TLC檢測反應終點,反應完後,加入4ml三乙胺中和至PH7-8,然後減壓濃縮,回收90-95%的有機溶劑。殘夜加800ml甲醇,降溫至0-5度,滴加40ml 36%的濃鹽酸,使其慢慢全溶,然後保溫於20-25度反應16-18小時,TLC確認反應終點,反應完後,減壓回收約650ml甲醇,然後冷卻,加入300ml純淨水,再將體系降溫至5-10度,攪拌析晶2-3小時,過濾,濾餅用少量 50%的甲醇水溶液洗滌,洗液和濾液合併,回收溶劑後排入廢水處理池,濾餅70度以下烘乾,得次甲基物粗品102.8g,HPLC含量97.2%,粗品用95%的乙醇重結晶,得次甲基物精品84.5g,HPLC含量99.2%,重量總收率84.5%。
C:6a-甲基氫化可的松的製備
在一個1000ml三口瓶中,加入100g 次甲基物、800ml乙醇,溶解後加入20g 5%的鈀碳,氮氣置換後保溫45-50度慢慢滴加200ml環己烯,約0.5小時滴完,然後升溫至55-60度繼續保溫攪拌反應1.5~2小時,TLC檢測反應終點,反應完後,放空,趁熱氮氣壓濾,50ml乙醇洗滌,濾餅送廠家回收;洗液和濾液合併,降溫至25-30度,慢慢滴加240ml 6N鹽酸,約0.5-1.0小時內滴完,然後再保溫於45-50度繼續轉位反應1.5-2.0小時,HPLC確認反應終點,反應完後,加入10%的燒鹼溶液中和至PH6.5-7.5,減壓濃縮90-95%的乙醇,降溫至20-25度,加入200ml自來水,再將體系降溫至5-10度,攪拌析晶2-3小時,過濾,洗滌,濾液與洗液濃縮回收溶劑後排入廢水處理池,濾餅100度以下烘乾,得6a-甲基氫化可的松粗品82.5g,HPLC含量98.3%,重量總收率82.5%;將上述6-甲基氫化可的松粗品用80%的乙醇水溶液重結晶,得6a-甲基氫化可的松精品72.8g,HPLC含量99.3%,本步重量總收率72.8%。
實例二
A、 醚化物的製備
在一個1000ml三口瓶中,加入100g 氫化可的松、200ml氯仿、80ml原甲酸三乙酯、2g濃硫酸,保溫於40-45度攪拌反應6~8小時,TLC檢測反應終點,反應完後,加入3ml 吡啶,攪拌20-25分鐘中和酸,然後減壓回收90-95%的有機溶劑,再加入500ml 50%的乙醇水溶液,將體系冷卻到-5-0度,攪拌結晶2~3小時,抽濾,少量乙醇水溶液洗滌,洗液和濾液合併,回收溶劑,濾餅70度以下烘乾,得醚化物101.2g,HPLC含量99.0%,重量收率101.2%。
B、次甲基物的製備
在一個1000ml三口瓶中,加入100g 醚化物、800ml四氫呋喃、50ml三氯甲烷,溶解後加入2g濃硫酸與50ml四氫呋喃的溶液 ,然後再加入70mlN-甲基苯胺與200ml 37%的甲醛溶液,升溫至25-30度保溫攪拌反應1.5~2.0小時,再在50-55度繼續反應1.0-1.5小時,TLC檢測反應終點,反應完後,加入6ml吡啶中和至PH7-8,然後減壓濃縮,回收90-95%的有機溶劑。殘夜加800ml甲醇,降溫至0-5度,滴加40ml 36%的濃鹽酸,使其慢慢全溶,然後保溫於20-25度反應16-18小時,TLC確認反應終點,反應完後,減壓回收約650ml甲醇,然後冷卻,加入300ml純淨水,再將體系降溫至5-10度,攪拌析晶2-3小時,過濾,濾餅用少量 50%的甲醇水溶液洗滌,洗液和濾液合併,回收溶劑後排入廢水處理池,濾餅70度以下烘乾,得次甲基物粗品101.2g,HPLC含量97.5%,粗品用95%的乙醇重結晶,得次甲基物精品82.6g,HPLC含量99.4%,重量總收率82.6%。
C、6a-甲基氫化可的松的製備
在一個1000ml三口瓶中,加入100g 次甲基物、800ml氯仿,溶解後加入20g 5%的鈀碳,氮氣置換後保溫45-50度慢慢滴加200ml環己烯,約0.5小時滴完,然後升溫至55-60度繼續保溫攪拌反應1.5~2小時,TLC檢測反應終點,反應完後,放空,趁熱氮氣壓濾,50ml氯仿洗滌,濾餅送廠家回收;洗液和濾液合併,降溫至25-30度,慢慢滴加240ml 6N鹽酸,約0.5-1.0小時內滴完,然後再保溫於45-50度繼續轉位反應1.5-2.0小時,HPLC確認反應終點,反應完後,加入10%的燒鹼溶液中和至PH6.5-7.5,減壓濃縮回收90-95%的氯仿,降溫至20-25度,加入200ml自來水,再將體系降溫至5-10度,攪拌析晶2-3小時,過濾,洗滌,濾液與洗液濃縮回收溶劑後排入廢水處理池,濾餅100度以下烘乾,得6a-甲基氫化可的松粗品81.5g,HPLC含量98.5%,重量總收率81.5%;將上述6a-甲基氫化可的松粗品用80%的乙醇水溶液重結晶,得6a-甲基氫化可的松精品70.8g,HPLC含量99.5%,本步重量總收率70.8%。
實施例三
A、 醚化物的製備
在一個1000ml三口瓶中,加入100g 氫化可的松、200ml甲苯、80ml原甲酸三乙酯、2gHCl與20ml乙醇的混合溶液,保溫於40-45度攪拌反應6~8小時,TLC檢測反應終點,反應完後,加入3ml 吡啶,攪拌20-25分鐘中和酸,然後減壓回收90-95%的有機溶劑,再加入500ml 50%的乙醇水溶液,將體系冷卻到-5-0度,攪拌結晶2~3小時,抽濾,少量乙醇水溶液洗滌,洗液和濾液合併,回收溶劑,濾餅70度以下烘乾,得醚化物100.8g,HPLC含量99.5%,重量收率100.8%。
B、次甲基物的製備
在一個1000ml三口瓶中,加入100g 醚化物、800ml甲苯、50ml三氯甲烷,溶解後加入2g對甲苯磺酸與50ml甲苯的混合溶液 ,然後再加入70mlN-甲基苯胺與200ml 37%的甲醛溶液,升溫至25-30度保溫攪拌反應1.5~2.0小時,再在50-55度繼續反應1.0-1.5小時,TLC檢測反應終點,反應完後,加入4ml三乙胺中和至PH7-8,然後減壓濃縮,回收90-95%的有機溶劑。殘夜加800ml甲醇,降溫至0-5度,滴加40ml 36%的濃鹽酸,使其慢慢全溶,然後保溫於20-25度反應16-18小時,TLC確認反應終點,反應完後,減壓回收約650ml甲醇,然後冷卻,加入300ml純淨水,再將體系降溫至5-10度,攪拌析晶2-3小時,過濾,濾餅用少量 50%的甲醇水溶液洗滌,洗液和濾液合併,回收溶劑後排入廢水處理池,濾餅70度以下烘乾,得次甲基物粗品100.8g,HPLC含量98.5%,粗品用95%的乙醇重結晶,得次甲基物精品82.2g,HPLC含量99.6%,重量總收率82.2%。
C、6a-甲基氫化可的松的製備
在一個1000ml三口瓶中,加入100g 次甲基物、800ml乙酸乙酯,溶解後加入20g 5%的鈀碳,氮氣置換後保溫45-50度慢慢滴加200ml環己烯,約0.5小時滴完,然後升溫至55-60度繼續保溫攪拌反應1.5~2小時,TLC檢測反應終點,反應完後,放空,趁熱氮氣壓濾,50ml乙酸乙酯洗滌,濾餅送廠家回收;洗液和濾液合併,降溫至25-30度,慢慢滴加240ml 6N鹽酸,約0.5-1.0小時內滴完,然後再保溫於45-50度繼續轉位反應1.5-2.0小時,HPLC確認反應終點,反應完後,加入10%的燒鹼溶液中和至PH6.5-7.5,減壓濃縮90-95%的乙酸乙酯,降溫至20-25度,加入200ml自來水,再將體系降溫至5-10度,攪拌析晶2-3小時,過濾,洗滌,濾液與洗液濃縮回收溶劑後排入廢水處理池,濾餅100度以下烘乾,得6a-甲基氫化可的松粗品84.2g,HPLC含量98.5%,重量總收率84.2%;將上述6-甲基氫化可的松粗品用80%的乙醇水溶液重結晶,得6a-甲基氫化可的松精品74.0g,HPLC含量99.5%,本步重量總收率74.0%。