一種氮雜萘醯亞胺類Cd2+探針分子及其合成方法與應用與流程
2023-05-16 23:55:56 3
本發明涉及Cd2+探針分子及其合成與應用。
背景技術:
Cd2+是一種非常有毒的金屬離子,是環境的主要金屬汙染物,對農作物及人體都有毒害作用。對作物來說,輕者使其體內的代謝過程發生紊亂,使作物生長受到抑制,重則導致作物死亡;對人來說,長期接觸Cd2+會導致腎功能損害、心血管疾病、鈣代謝紊亂、嗜酸性粒細胞增多症和某些癌症等疾病。由於Cd2+廣泛應用於電池中,空氣、水、土壤中Cd2+的汙染較為嚴重,因此用於識別和檢測環境樣品以及活細胞內Cd2+含量的探針就顯得尤其重要。
然而,正如在《化學評論》(Chem.Rev.)的2014年第114卷4564-4601頁中所提到的,目前已報導的Cd2+分子探針在檢測Cd2+時易受到結合性能相近的Zn2+的幹擾,抗Zn2+幹擾性較差。而由於Zn2+在人體內普遍存在,這給人體內Cd2+的檢測帶來諸多不便。為此,開發不受Zn2+幹擾的Cd2+螢光探針仍然是目前研究的重點。
技術實現要素:
本發明是要解決現有的檢測Cd2+的分子探針易受Zn2+幹擾、抗Zn2+幹擾性較差的技術問題,從而提供一種氮雜萘醯亞胺類Cd2+探針分子及其合成方法與應用。
本發明的氮雜萘醯亞胺類Cd2+探針分子,其結構式為:
其中,所述的R為C1~C4直鏈烷基。
上述氮雜萘醯亞胺類Cd2+探針分子的合成方法,按以下步驟進行:
一、將吡喃並[3,4,5-de]喹啉-2,4,6(1H)-三酮與烷基胺以摩爾比為1:(4.8~5.2)的比例加入至乙醇中,在氮氣保護下加熱至沸騰回流4~6h;將反應液旋幹,再經過矽膠柱色譜分離,得到中間體Ⅰ;
二、將步驟一所得中間體Ⅰ與三氯氧磷以摩爾比為1:(9.5~10.5)的比例加入至二氧六環中,在75~85℃的條件下反應2~3h;再用氨水將反應液的pH值調至鹼性;將反應液旋幹,再經過矽膠柱色譜分離,得到中間體Ⅱ;
三、將步驟二所得中間體Ⅱ與2-氨基苄胺以摩爾比為1:(4.8~5.2)的比例加入至乙二醇甲醚中,在氮氣保護下加熱至沸騰回流6~8h;將反應液旋幹,再經過矽膠柱色譜分離,得到中間體Ⅲ;
四、將步驟三所得中間體Ⅲ、吡啶溶於二氯甲烷中,滴加2-氯乙醯氯,其中吡啶的物質的量為中間體Ⅲ的1.4~2.0倍,2-氯乙醯氯的物質的量為中間體Ⅲ的1.1~1.3倍,室溫下反應2~3h;將反應液旋幹,再經過矽膠柱色譜分離,得到中間體Ⅳ;
五、將步驟四所得中間體Ⅳ、二(2-吡啶甲基)胺、N,N-二異丙基乙基胺以摩爾比為1:(4.8~5.2):(6~10)的比例加入至乙腈中,在氮氣保護下加熱至沸騰回流6~8h;將反應液旋幹,再經過矽膠柱色譜分離,得到氮雜萘醯亞胺類Cd2+探針分子。
上述的氮雜萘醯亞胺類Cd2+探針分子的應用,是用氮雜萘醯亞胺類Cd2+探針分子對溶液或細胞內的Cd2+進行螢光檢測。
本發明的氮雜萘醯亞胺類Cd2+探針分子可直接用於在水、甲醇、DMSO、DMF溶劑或它們的混合溶劑中Cd2+的螢光增強檢測,也可對生物組織、細胞微環境中Cd2+進行螢光成像檢測,且不受Zn2+幹擾,具有較高的選擇性和靈敏度。而且在螢光檢測時,該類探針分子的激發波長位於可見光區,避免了紫外光的應用。該類探針分子可用於環境樣品、生物組織以及活細胞微環境中Cd2+的螢光檢測,具有廣泛的潛在應用價值。
附圖說明
圖1為實施例1製備的氮雜萘醯亞胺類Cd2+探針分子在加入金屬離子前後的螢光光譜變化圖;
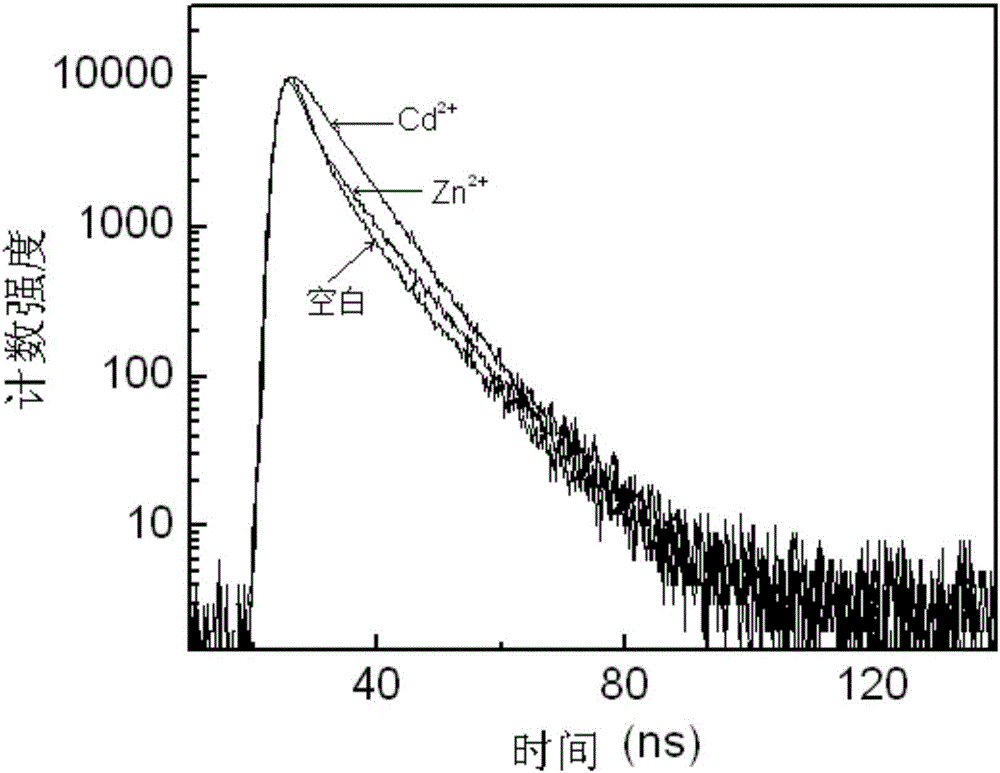
圖2為實施例1製備的氮雜萘醯亞胺類Cd2+探針分子在加入Cd2+和Zn2+前後的螢光衰減曲線;
圖3為實施例1製備的氮雜萘醯亞胺類Cd2+探針分子對不同濃度的Cd2+的螢光光譜變化圖;
圖4為實施例1製備的氮雜萘醯亞胺類Cd2+探針分子對不同濃度的Cd2+的螢光強度變化曲線;
圖5為實施例1中,酵母細胞放到混合液中培養後的明場照片;
圖6為實施例1中,酵母細胞放到混合液中培養後的螢光照片;
圖7為實施例1中,染色的酵母細胞放到含Cd2+的溶液中培養後的明場照片;
圖8為實施例1中,染色的酵母細胞放到含Cd2+的溶液中培養後的螢光照片。
具體實施方式
具體實施方式一:本實施方式的氮雜萘醯亞胺類Cd2+探針分子,其結構式為:
其中,所述的R為C1~C4直鏈烷基。
具體實施方式二:具體實施方式一所述的氮雜萘醯亞胺類Cd2+探針分子的合成方法,按以下步驟進行:
一、將吡喃並[3,4,5-de]喹啉-2,4,6(1H)-三酮與烷基胺以摩爾比為1:(4.8~5.2)的比例加入至乙醇中,在氮氣保護下加熱至沸騰回流4~6h;將反應液旋幹,再經過矽膠柱色譜分離,得到中間體Ⅰ;
二、將步驟一所得中間體Ⅰ與三氯氧磷以摩爾比為1:(9.5~10.5)的比例加入至二氧六環中,在75~85℃的條件下反應2~3h;再用氨水將反應液的pH值調至鹼性;將反應液旋幹,再經過矽膠柱色譜分離,得到中間體Ⅱ;
三、將步驟二所得中間體Ⅱ與2-氨基苄胺以摩爾比為1:(4.8~5.2)的比例加入至乙二醇甲醚中,在氮氣保護下加熱至沸騰回流6~8h;將反應液旋幹,再經過矽膠柱色譜分離,得到中間體Ⅲ;
四、將步驟三所得中間體Ⅲ、吡啶溶於二氯甲烷中,滴加2-氯乙醯氯,其中吡啶的物質的量為中間體Ⅲ的1.4~2.0倍,2-氯乙醯氯的物質的量為中間體Ⅲ的1.1~1.3倍,室溫下反應2~3h;將反應液旋幹,再經過矽膠柱色譜分離,得到中間體Ⅳ;
五、將步驟四所得中間體Ⅳ、二(2-吡啶甲基)胺、N,N-二異丙基乙基胺以摩爾比為1:(4.8~5.2):(6~10)的比例加入至乙腈中,在氮氣保護下加熱至沸騰回流6~8h;將反應液旋幹,再經過矽膠柱色譜分離,得到氮雜萘醯亞胺類Cd2+探針分子。
具體實施方式三:本實施方式與具體實施方式二不同的是:步驟一中吡喃並[3,4,5-de]喹啉-2,4,6(1H)-三酮、烷基胺以摩爾比為1:5。其它與具體實施方式二相同。
具體實施方式四:本實施方式與具體實施方式二或三不同的是:步驟一中在氮氣保護下加熱至沸騰回流5h。其它與具體實施方式二或三相同。
具體實施方式五:本實施方式與具體實施方式二至四之一不同的是步驟二中中間體Ⅰ與三氯氧磷以摩爾比為1:10。其它與具體實施方式二至四之一相同。
具體實施方式六:本實施方式與具體實施方式二至五之一不同的是步驟二中將反應液的pH值調至7.5~8.5。其它與具體實施方式二至五之一相同。
具體實施方式七:本實施方式與具體實施方式二至六之一不同的是步驟四中吡啶的物質的量為中間體Ⅲ的1.4倍,2-氯乙醯氯的物質的量為中間體Ⅲ的1.2倍。其它與具體實施方式二至六之一相同。
具體實施方式八:本實施方式與具體實施方式二至七之一不同的是步驟五中中間體Ⅳ、二(2-吡啶甲基)胺、N,N-二異丙基乙基胺的摩爾比為1:5:10。其它與具體實施方式二至七之一相同。
具體實施方式九:本實施方式一所述的氮雜萘醯亞胺類Cd2+探針分子的應用,是用氮雜萘醯亞胺類Cd2+探針分子對溶液或細胞內的Cd2+進行螢光增強檢測。
用以下的實施例驗證本發明的有益效果:
實施例1:本實施例的氮雜萘醯亞胺類Cd2+探針分子的合成方法,按以下步驟進行:
一、將吡喃並[3,4,5-de]喹啉-2,4,6(1H)-三酮(450mg,2.09mmol)、正丁胺(764mg,10.45mmol)加入至50mL乙醇中,在氮氣保護下回流6h,停止反應;將反應液旋幹,以二氯甲烷-乙酸乙酯混合液為洗脫液,矽膠柱色譜分離,得中間體Ⅰ,產量328mg,收率為58%,熔點:264.8~265.2℃。將得到的中間體Ⅰ進行分析,結果如下:1H NMR(CDCl3,600MHz):δ8.18(d,J=7.2Hz,1H),7.79(t,J=8.1Hz,1H),7.72(d,J=8.4Hz,1H),7.70(s,1H),4.14(CH2CH2CH2CH3,t,J=7.5Hz,2H),1.69(CH2CH2CH2CH3,m,2H),1.44(CH2CH2CH2CH3,m,2H),0.99(CH3,t,J=7.2Hz,3H)ppm.13C NMR(CDCl3,150MHz):δ163.96,162.85,161.84,137.51,133.86,131.92,125.70,124.88,123.18,120.74,115.27,40.75,30.07,20.33,13.78ppm.HRMS m/z(TOF MS ES+):calcd for C15H15N2O3+(M+H+)271.1077,found 271.1076。從分析結果可知,中間體Ⅰ的結構式為步驟一中利用吡喃並[3,4,5-de]喹啉-2,4,6(1H)-三酮合成了中間體Ⅰ,即
二、將300mg步驟一所得的中間體Ⅰ(1.11mmol)、1.68g三氯氧磷(11.10mmol)加入至30mL二氧六環中,在80℃下反應3h,停止反應;用氨水將反應液調至pH值為8;將反應液旋幹,以二氯甲烷為洗脫液,矽膠柱色譜分離,得中間體Ⅱ,其產量297mg,收率為93%,熔點:136.2~136.4℃。將中間體Ⅱ進行分析,結果如下:1H NMR(CDCl3,600MHz):δ8.58(d,J=7.2Hz,1H),8.36(d,J=8.4Hz,1H),8.27(s,1H),7.99(t,J=7.8Hz,1H),4.16(CH2CH2CH2CH3,t,J=7.8Hz,2H),1.69(CH2CH2CH2CH3,m,2H),1.43(CH2CH2CH2CH3,m,2H),0.97(CH3,t,J=7.5Hz,3H)ppm.13C NMR(CDCl3,150MHz):δ162.53,161.94,152.54,146.90,133.94,132.27,131.55,131.03,124.20,122.97,121.55,40.65,30.10,20.30,13.78ppm.HRMS m/z(TOF MS ES+):calcd for C15H14ClN2O2+(M+H+)289.0738,found 289.0740。從分析結果可知中間體Ⅱ的結構式為:
三、將280mg步驟二所得中間體Ⅱ(0.97mmol)與592mg 2-氨基苄胺(4.85mmol)加入至30mL乙二醇甲醚中,在氮氣保護下加熱至沸騰回流8h,停止反應;將反應液旋幹,以二氯甲烷-乙酸乙酯混合液為洗脫液,矽膠柱色譜分離,得中間體Ⅲ,其產量302mg,收率為83%,熔點:204.4~204.8℃。將中間體Ⅲ進行分析,結果如下:1H NMR(DMSO-d6,600MHz):δ8.07(NHCH2,t,J=5.7Hz,1H),8.00(d,J=8.4Hz,1H),7.90(d,J=9.0Hz,1H),7.76(t,J=7.8Hz,1H),7.69(s,1H),7.11(d,J=9.0Hz,1H),6.97(t,J=8.4Hz,1H),6.65(d,J=9.0Hz,1H),6.52(t,J=7.8Hz,1H),5.17(NH2,s,2H),4.54(NHCH2,d,J=5.4Hz,2H),3.99(CH2CH2CH2CH3,t,J=7.5Hz,2H),1.58(CH2CH2CH2CH3,m,2H),1.34(CH2CH2CH2CH3,m,2H),0.92(CH3,t,J=7.2Hz,3H)ppm.13C NMR(DMSO-d6,150MHz):δ163.57,163.20,157.98,147.22,146.98,130.97,130.75,130.30,129.56,128.30,124.07,122.71,122.53,117.93,116.46,116.32,115.29,41.51,40.52,30.04,20.25,14.18ppm.HRMSm/z(TOF MS ES+):calcd for C22H23N4O2+(M+H+)375.1816,found 375.1815。從分析結果可知,中間體Ⅲ的結構式為:
四、將150mg步驟三所得中間體Ⅲ(0.40mmol)、44mg吡啶(0.56mmol)溶於10mL二氯甲烷中,滴加54mg 2-氯乙醯氯(0.48mmol),室溫下反應2h,停止反應;將反應液旋幹,以二氯甲烷-乙酸乙酯混合液為洗脫液,矽膠柱色譜分離,得到中間體Ⅳ;其產量為110mg,收率為61%,熔點為213.6-214.1℃。將中間體Ⅳ進行分析,結果如下:1H NMR(DMSO-d6,600MHz):δ9.96(CONH,s,1H),8.20(NHCH2,t,J=5.1Hz,1H),8.01(d,J=7.2Hz,1H),7.90(d,J=8.4Hz,1H),7.70(t,J=7.8Hz,1H),7.72(s,1H),7.47(d,J=7.8Hz,1H),7.40(d,J=7.2Hz,1H),7.28(t,J=7.5Hz,1H),7.19(t,J=7.5Hz,1H),4.68(NHCH2,d,J=5.4Hz,2H),3.35(COCH2,s,2H),3.99(CH2CH2CH2CH3,t,J=7.5Hz,2H),1.59(CH2CH2CH2CH3,m,2H),1.34(CH2CH2CH2CH3,m,2H),0.92(CH3,t,J=7.2Hz,3H)ppm.13C NMR(DMSO-d6,150MHz):δ169.07,165.60,163.54,163.15,157.75,135.56,133.45,130.85,130.50,128.86,128.81,127.96,127.77,126.32,125.80,124.34,122.77,118.06,43.75,41.95,40.54,30.04,20.24,14.17.HRMS m/z(TOF MS ES+):calcd for C24H24ClN4O3+(M+H+)451.1531,found451.1532。從分析結果可知,中間體Ⅳ的結構式為:
五、將50mg步驟四所得中間體Ⅳ(0.11mmol))、110mg二(2-吡啶甲基)胺(0.55mmol)、142mg N,N-二異丙基乙基胺(1.10mmol)加入至30mL乙腈中,在氮氣保護下加熱至沸騰回流8h;將反應液旋幹,以二氯甲烷-甲醇混合液為洗脫液,矽膠柱色譜分離,得到氮雜萘醯亞胺類Cd2+探針分子。其產量為55mg,收率為82%,熔點為165.8~166.1℃。將氮雜萘醯亞胺類Cd2+探針分子進行分析,結果如下:1H NMR(DMSO-d6,600MHz):δ10.14(CONH,s,1H),8.50(d,J=4.2Hz,2H),8.22(NHCH2,t,J=5.4Hz,1H),8.00(d,J=7.2Hz,1H),7.85(d,J=8.4Hz,1H),7.74(m,3H),7.66(t,J=8.1Hz,2H),7.40(m,3H),7.26(t,J=7.5Hz,1H),7.20(t,J=6.3Hz,2H),7.13(t,J=7.5Hz,1H),4.78(NHCH2,d,J=5.4Hz,2H),3.98(CH2CH2CH2CH3,t,J=7.5Hz,2H),3.84(N(CH2Py)2,s,4H),3.43(COCH2,s,2H),1.58(CH2CH2CH2CH3,m,2H),1.33(CH2CH2CH2CH3,m,2H),0.92(CH3,t,J=7.2Hz,3H)ppm.13C NMR(DMSO-d6,150MHz):δ169.66,163.54,163.19,158.43,157.91,149.51,147.23,137.23,137.03,136.40,131.31,131.21,130.73,130.27,128.87,127.78,125.16,124.30,123.87,123.61,122.80,122.72,118.07,60.04,58.12,41.36,40.55,30.05,20.24,14.18ppm.HRMS m/z(TOF MS ES+):calcd for C36H36N7O3+(M+H+)614.2874,found 614.2883。從分析結果可知,氮雜萘醯亞胺類Cd2+探針分子的結構式為:
按三羥甲基氨基甲烷的濃度為0.01mol/L、實施例1製備的氮雜萘醯亞胺類Cd2+探針分子的濃度為1.0×10-5mol/L依次將三羥甲基氨基甲烷、氮雜萘醯亞胺類Cd2+探針分子加入到體積比為1:1的甲醇和水混合液中,用鹽酸將溶液pH調至7.20,分別加入各種常見的金屬離子Na+、K+、Mg2+、Ca2+、Cr3+、Fe3+、Co2+、Ni2+、Cu2+、Zn2+、Ag+、Cd2+、Hg2+、Al3+,金屬離子的濃度為探針濃度的5倍,測定其螢光光譜,得到的螢光光譜圖如圖1所示,從圖1可以看出,空白探針分子在543nm處的螢光較弱,螢光量子產率為0.04。僅Cd2+導致探針分子在543nm處的螢光顯著增強,其螢光量子產率增加約3.3倍;而Zn2+導致探針分子在543nm處的螢光略有猝滅。由此可見本實施例的氮雜萘醯亞胺類Cd2+探針分子可高選擇性螢光增強識別Cd2+,並且可以應用螢光增強信號區分Cd2+和Zn2+,從避免了Cd2+檢測過程中Zn2+的幹擾。
按三羥甲基氨基甲烷的濃度為0.01mol/L、實施例1製備的氮雜萘醯亞胺類Cd2+探針分子的濃度為1.0×10-5mol/L依次將三羥甲基氨基甲烷、氮雜萘醯亞胺類Cd2+探針分子加入到體積比為1:1的甲醇和水混合液中,用鹽酸將溶液pH調至7.20,分別加入Cd2+和Zn2+,金屬離子的濃度為探針濃度的5倍,測定發射波長在543nm處的螢光衰減曲線如圖2所示,從圖2計算可得,空白探針分子的螢光壽命為6.20ns,加入Cd2+後探針分子的螢光壽命增加至8.87ns,而加入Zn2+後探針分子的螢光壽命為7.18ns。由此可見本實施例的氮雜萘醯亞胺類Cd2+探針分子可以用螢光壽命區分Cd2+和Zn2+。
按三羥甲基氨基甲烷的濃度為0.01mol/L、實施例1製備的氮雜萘醯亞胺類Cd2+探針分子的濃度為1.0×10-5mol/L依次將三羥甲基氨基甲烷、氮雜萘醯亞胺類Cd2+探針分子加入到體積比為1:1的甲醇和水混合液中,用鹽酸將溶液pH調至7.20,加入Cd2+,Cd2+的濃度為0~1.0×10-5mol/L,研究不同Cd2+溶度對探針分子螢光光譜的影響,得到的螢光光譜如圖3所示,從圖3可以看出,隨著Cd2+的加入,探針分子在543nm處的螢光強度逐漸上升,當Cd2+的濃度為1.0×10-5mol/L時增強約3.5倍;而當Cd2+濃度超過1.0×10-5mol/L時,其螢光強度基本保持不變。探針分子螢光強度與Cd2+濃度的關係曲線如圖4所示,從圖4可以計算得出,探針分子對Cd2+的檢出限為1.08×10-8mol/L,在Cd2+濃度1.08×10-8-1.0×10-5mol/L範圍內,探針分子的螢光強度與Cd2+濃度呈良好的線性關係,由此可見本實施例製備的氮雜萘醯亞胺類Cd2+探針分子可以定量檢測Cd2+。
按三羥甲基氨基甲烷的濃度為0.01mol/L、實施例1製備的氮雜萘醯亞胺類Cd2+探針分子的濃度為4.0×10-5mol/L依次將三羥甲基氨基甲烷、氮雜萘醯亞胺類Cd2+探針分子加入到體積比為1:1的DMSO和水混合液中,用鹽酸將溶液pH調至7.20,得到混合液。酵母細胞在混合液中培養染色0.5h後進行明場和螢光成像,得到的明場照片如圖5所示,得到的螢光照片如圖6所示,由圖6可知,細胞發出微弱的黃綠色螢光。將染色後的酵母細胞加入至Cd2+濃度為4.0×10-5mol/L的溶液中培養0.5h,進行明場和螢光成像,得到的明場照片如圖7所示,得到的螢光照片如圖8所示,由圖8可知,染色後的酵母細胞在含Cd2+的溶液中培養後細胞發出較強的黃綠色螢光,表明在細胞內探針分子對Cd2+具有較高的檢測性能。