多肽修飾的低聚原花青素及其製備方法與應用與流程
2023-05-04 07:56:32 3
本發明屬於牙釉質原位修復材料及牙科植入體表面抗菌材料領域,特別涉及多肽修飾的低聚原花青素及其製備方法與應用。
背景技術:
齲齒是一種細菌引起的疾病,主要表現為牙齒的脫礦現象。目前,用於填充齲齒患處的材料包括金屬材料、陶瓷材料和樹脂材料,然而由於牙齒基質與填充材料之間性質差異巨大,兩者界面間相互作用力不強,50%~60%的填充材料在首次治療後不久脫落(L.L.Kirkevang,M.A.Wenzel,Caries Res.43(2009)286)。近年來,誘導牙釉質定點再礦化的材料得到了廣泛的研究和關注,這類材料通過模擬有機基質誘導牙釉質再礦化,目前可用的這類材料包括生物大分子、脂質體、自組裝膜等。
葡萄籽提取物(GSE)是從天然葡萄籽中提取的混合物,主要含多酚類化合物、萜類化合物、維生素、胺基酸、生物鹼等多種生物活性物質,能抑制致齲菌的生長和粘附。施桔紅等以原花青素含量為61.6%的葡萄籽提取物誘導受損牙本質再礦化,將經過脫礦處理的牙本質塊進行pH循環處理8天,以考察GSE對牙本質再礦化的促進作用,結果表明10%的GSE能促進脫礦牙本質的再礦化作用(施桔紅,黎紅,王伊娜.葡萄籽提取物促進早期牙本質齲損再礦化作用的體外研究.[J]上海口腔醫.2015,24(1).)。雖然GSE能促進脫礦牙本質的再礦化,但GSE具有該作用主要是因為其中含有的原花青素能減緩牙本質上膠原的生物降解,起到穩定膠原的作用有關,從而為礦物質的沉積提供網狀支撐。與牙本質相比,牙釉質中的膠原等有機成分的含量明顯更低,且原花青素對作為牙釉質主要無機成分的對羥基磷灰石沒有吸附作用,因而原花青素無法牢固地作用於牙釉質上,難以用於牙釉質的修復。
基於以上技術現狀,若能對原花青素的結構進行改進,並利用原花青素能抑制致齲菌的生長和粘附的特點,開發出對牙釉質具有良好的吸附效果、能有效促進牙釉質再礦化和具有抑菌作用的誘導牙齒定點再礦化的材料,對於齲齒修復和牙科植入體表面抗菌都將產生重要的意義。
技術實現要素:
本發明的目的在於克服現有技術的不足,提供多肽修飾的低聚原花青素及其製備方法,並證明所述多肽修飾的低聚原花青素可作為牙釉質原位再礦化誘導劑以及牙科植入體表面抗菌材料應用。
本發明提供的多肽修飾的低聚原花青素,結構式如式(Ⅰ)所示:
式(Ⅰ)中,R代表結構式如式(Ⅱ)所示的低聚原花青素分子中的任意一個R1被去掉後形成的基團,式(Ⅱ)中,n=0、1、2或3(即低聚原花青素為二聚、三聚、四聚或五聚原花青素),R1為羥基,
本發明提供的上述多肽修飾的低聚原花青素的製備方法,步驟如下:
(1)丙烯醯基修飾的低聚原花青素的合成
①配製第一種溶液
將低聚原花青素溶解於二甲基甲醯胺中形成第一種溶液,所述低聚原花青素的聚合度為2、3、4或5,低聚原花青素在第一種溶液中的濃度為10~40mg/mL;
②配製第二種溶液
將三乙胺溶解於第一種溶液中形成第二種溶液,所述三乙胺與第一種溶液中低聚原花青素的摩爾比為(1~1.5):1;
③合成
在氬氣保護條件下於-5~5℃將丙烯醯氯加入到第二種溶液中,在室溫反應至少24h,然後經透析、冷凍乾燥,即得丙烯醯基修飾的低聚原花青素;所述丙烯醯氯與第二種溶液中低聚原花青素的摩爾比為(1~1.5):1;
(2)帶保護基團的多肽修飾的低聚原花青素的合成
①配製第三種溶液
將步驟(1)製備的丙烯醯基修飾的低聚原花青素溶解於去離子水中形成第三種溶液,所述去離子水的量以所述丙烯醯基修飾的低聚原花青素能在去離子水中完全溶解為限;
②配製第四種溶液
以結構式如式(Ⅲ)所示的多肽為溶質,將所述溶質溶解於去離子水中形成第四種溶液,所述去離子水的量以溶質能在去離子水中完全溶解為限,
③合成
將二甲基苯基磷加入第三種溶液中並混合均勻,然後加入第四種溶液並混合均勻,在密封條件下於室溫反應至少24h,經透析、冷凍乾燥,即得帶保護基團的多肽修飾的低聚原花青素;
所述第四種溶液的加入量應使第四種溶液中的巰基與第三種溶液中的碳碳雙鍵的摩爾比為(1~1.5):1,所述二甲基苯基磷與第四種溶液中的巰基的摩爾比為(10-6~10-5):1;
(3)多肽修飾的低聚原花青素的合成
①配製第五種溶液
將氨水、二氧六環、濃度為3~5mol/L的氫氧化鈉水溶液混合均勻得到第五種溶液,第五種溶液中,氨水、二氧六環、氫氧化鈉的摩爾比為(1.9~2.1):(0.9~1):(0.08~0.12);
②合成
以步驟(2)製備的帶保護基團的多肽修飾的低聚原花青素為溶質,將所述溶質溶解於第五種溶液中並混合均勻,在密封條件下於室溫反應至少24h,然後經透析、冷凍乾燥,即得多肽修飾的低聚原花青素;所述第五種溶液的量以溶質能在第五種溶液中完全溶解為限。
本發明提供了上述多肽修飾的低聚原花青素作為牙釉質原位再礦化誘導劑以及牙科植入體表面抗菌材料的應用。實驗表明:本發明所述多肽修飾的低聚原花青素對羥基磷灰石具有牢固的吸附作用,並能在羥基磷灰表面吸附聚集成膜;本發明所述多肽修飾的低聚原花青素能在模擬唾液中誘導羥基磷灰石在磨損牙釉質塊表面沉積和結晶,具有促進牙釉質再礦化的能力,並且,將本發明所述多肽修飾的低聚原花青素與三價鐵離子配合使用,能進一步提升所述多肽修飾的低聚原花青素在模擬唾液中對牙釉質再礦化的能力,說明三價鐵離子可與本發明所述多肽修飾的低聚原花青素配合作為牙釉質原位再礦化誘導劑應用。本發明所述多肽修飾的低聚原花青素能有效防止細菌(包括死細菌和活細菌)在牙釉質上粘附,阻止粘附的細菌在其上繁殖,從而體現出抑菌作用,基於該特性,本發明所述多肽修飾的低聚原花青素可作為牙科植入體表面抗菌材料應用。
應用時,將所述多肽修飾的低聚原花青素配製成水溶液使用,所述水溶液中,多肽修飾的低聚原花青素的濃度為1000~3000ppm。
與現有技術相比,本發明具有以下有益效果:
1.本發明提供了一種新型結構的誘導牙釉質定點再礦化的材料—多肽修飾的低聚原花青素,豐富了牙釉質定點再礦化的材料的種類。
2.實驗表明:本發明所述多肽修飾的低聚原花青素對羥基磷灰石具有牢固的吸附作用,並能在羥基磷灰表面吸附聚集成膜,本發明所述多肽修飾的低聚原花青素能在模擬唾液中誘導羥基磷灰石在磨損牙釉質塊表面沉積和結晶,具有促進牙釉質再礦化的能力,並且,與三價鐵離子配合使用,能進一步提升所述多肽修飾的低聚原花青素在模擬唾液中對牙釉質再礦化的能力,可在作為牙釉質原位再礦化誘導劑中應用(見實施例5~9);本發明所述多肽修飾的低聚原花青素能有效防止細菌(包括死細菌和活細菌)在牙釉質上粘附,阻止粘附的細菌在其上繁殖,從而體現出抑菌作用,可作為牙科植入體表面抗菌材料應用(見實施例10)。
3.本發明所述多肽修飾的低聚原花青素在應用時配製成多肽修飾的低聚原花青素的濃度為1000~3000ppm的水溶液直接浸泡進行再礦化的部位即可吸附在相應部位,在吸附後可誘導相應部位再礦化,應用方式簡單,具有簡化治療操作和降低治療成本的優勢,容易在實際應用中推廣。
4.本發明還提供了上述多肽修飾的低聚原花青素的製備方法,該方法採用常規原料及設備即可實現,易於實現工業化生產。
附圖說明
圖1是本發明所述多肽修飾的低聚原花青素的合成過程示意圖;
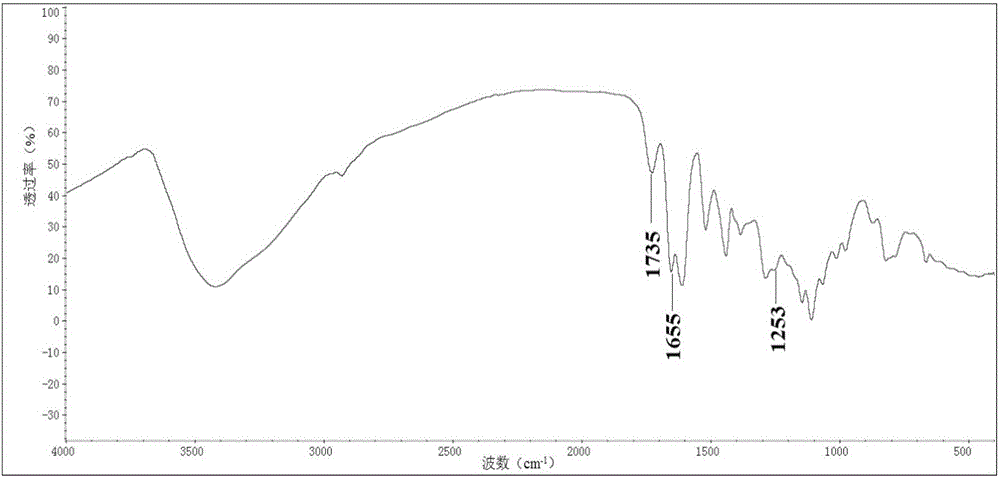
圖2是實施例1製備的丙烯醯基修飾的二聚原花青素的紅外譜圖;
圖3是實施例1製備的丙烯醯基修飾的二聚原花青素的核磁氫譜;
圖4是實施例1製備的帶保護基團的多肽修飾的二聚原花青素的紅外譜圖;
圖5是實施例1製備的多肽修飾的二聚原花青素的飛行時間質譜;
圖6是實施例5中各樣品的螢光分布圖,其中,圖(A)~(C)分別為羅丹明標記的SAP-2-OPC在酸蝕牙釉質塊上、羅丹明標記的SAP-2-OPC在未酸蝕牙釉質塊上、羅丹明標記的二聚原花青素在未酸蝕牙釉質塊上的螢光分布圖;
圖7是實施例6中不同濃度的SAP-2-OPC在羥基磷灰石粉末上的吸附量圖;
圖8是實施例7中SAP-2-OPC在羥基磷灰石片表面聚集成膜後的SEM照片;
圖9是實施例8的實驗組中礦化不同時間的酸蝕牙釉質塊表面的SEM圖,其中,圖(A)、(B)的礦化時間分別為1天和2周。
圖10是實施例8的空白組中礦化不同時間的酸蝕牙釉質塊表面的SEM圖,其中,圖(A)、(B)的礦化時間分別為1天和2周。
圖11是實施例9的實驗組中礦化不同時間的酸蝕牙釉質塊表面的SEM圖,其中,圖(A)、(B)的礦化時間分別為1天和2周。
圖12是實施例10實驗組和空白組在波長600nm處測吸光度值柱狀圖;
圖13是實施例10中實驗組和空白組的羥基磷灰石片表面的SEM照片,其中,圖(A)、(B)為實驗組的SEM照片,圖(C)、(D)為空白組的SEM照片;
圖14是實施例10中空白組與實驗組樣品的共聚焦顯微鏡圖,其中,圖(A)、(B)分別為空白組的樣品表面的活菌和死菌被染色後的共聚焦顯微鏡圖,圖(C)、(D)分別為實驗組的樣品表面的活菌和死菌被染色後的共聚焦顯微鏡圖。
具體實施方式
以下通過實施例並結合附圖對本發明所述多肽修飾的低聚原花青素及其製備方法與應用作進一步說明。
下述實施例1~4中,所述二聚、三聚、四聚和五聚原花青素均由兒茶素與兒茶素聚合而成;步驟(2)中使用的多肽購買自上海波泰生物科技有限公司,其結構式如式(Ⅲ)所示:
實施例1
本實施例中,提供多肽修飾的二聚原花青素的製備方法,其合成路線如圖1所示,步驟如下:
(1)丙烯醯基修飾的二聚原花青素的合成
①配製第一種溶液
在室溫、常壓條件下將0.59g二聚原花青素溶解於40mL二甲基甲醯胺中形成第一種溶液,二聚原花青素在第一種溶液中的濃度為14.8mg/mL;
②配製第二種溶液
在室溫、常壓條件下將三乙胺(TEA)溶解於第一種溶液中形成第二種溶液,所述三乙胺與第一種溶液中二聚原花青素的摩爾比為1.1:1;
③合成
在0~5℃的冰水浴及氬氣保護條件下將丙烯醯氯(Acryloyl chloride)滴加到第二種溶液中,在室溫室溫、常壓條件下反應24h,然後在去離子水中用截留分子量為500道爾頓的透析袋透析1天,再冷凍乾燥,即得丙烯醯基修飾的二聚原花青素;所述丙烯醯氯與第二種溶液中二聚原花青素的摩爾比為1.1:1。
該步驟製備的丙烯醯基修飾的二聚原花青素紅外譜圖和核磁氫譜分別如圖2和圖3所示,圖2中,1735cm-1和1253cm-1處的峰分別表示酯基中C=0的伸縮振動和O-C-O的伸縮振動,1655cm-1處的峰代表C=C的伸縮振動,圖2中,7.95ppm和5.87ppm處的峰分別代表C=CH-O和-C=CH上的質子氫,峰面積比為1:2,圖2和圖3說明該步驟已經在二聚原花青素的基礎上引入了丙烯醯基。
(2)帶保護基團的多肽修飾的二聚原花青素的合成
①配製第三種溶液
在常壓、室溫條件下將步驟(1)製備的18.3mg丙烯醯基修飾的二聚原花青素溶解於10mL去離子水中形成第三種溶液;
②配製第四種溶液
以40mg結構式如式(Ⅲ)所示的多肽為溶質,在室溫、常壓條件下將所述溶質溶解於10mL去離子水中形成第四種溶液;
③合成
將二甲基苯基磷(Dimethylphenylphosphine)加入第三種溶液中,在室溫、常壓攪拌混合均勻,然後加入第四種溶液並攪拌混合均勻,在密封條件下於室溫、常壓反應24h,然後在去離子水中用截留分子量為1500道爾頓的透析袋透析1天,冷凍乾燥,即得帶保護基團的多肽修飾的二聚原花青素;
所述第四種溶液的加入量應使第四種溶液中的巰基與第三種溶液中的碳碳雙鍵的摩爾比為1.1:1,所述二甲基苯基磷與第四種溶液中的巰基的摩爾比為10-6:1。
該步驟製備的帶保護基團的多肽修飾的二聚原花青素的紅外譜圖如圖4所示,圖4中1550cm-1處的峰代表醯胺II帶,1680cm-1處的峰代表醯胺I帶,說明該步驟在步驟(1)製備的產物的基礎上引入了帶保護基團的多肽。
(3)多肽修飾的二聚原花青素的合成
①配製第五種溶液
在室溫、常壓條件下將氨水、二氧六環、濃度為4mol/L的氫氧化鈉水溶液混合均勻得到第五種溶液,第五種溶液中,氨水、二氧六環、氫氧化鈉的摩爾比為2:1:0.1;
②合成
以20mg步驟(2)製備的帶保護基團的多肽修飾的二聚原花青素為溶質,在室溫、常壓條件下將所述溶質溶解於1mL第五種溶液中並攪拌混合均勻,在密封條件下於室溫、常壓反應24h,然後然後在去離子水中用截留分子量為1000道爾頓的透析袋透析24h,冷凍乾燥,即得多肽修飾的二聚原花青素,記作SAP-2-OPC,其飛行時間質譜如圖5所示,圖5中,
1919.523為SAP-2-OPC的分子離子峰,SAP-2-OPC的結構式為:
實施例2
本實施例中,提供多肽修飾的三聚原花青素的製備方法,其合成路線如圖1所示,步驟如下:
(1)丙烯醯基修飾的三聚原花青素的合成
①配製第一種溶液
在室溫、常壓條件下將0.885g三聚原花青素溶解於40mL二甲基甲醯胺中形成第一種溶液,三聚原花青素在第一種溶液中的濃度為22mg/mL;
②配製第二種溶液
在室溫、常壓條件下將三乙胺溶解於第一種溶液中形成第二種溶液,所述三乙胺與第一種溶液中三聚原花青素的摩爾比為1.1:1;
③合成
在0~5℃的冰水浴及氬氣保護條件下將丙烯醯氯滴加到第二種溶液中,在室溫室溫、常壓條件下反應24h,然後在去離子水中用截留分子量為750道爾頓的透析袋透析1天,再冷凍乾燥,即得丙烯醯基修飾的三聚原花青素;所述丙烯醯氯與第二種溶液中三聚原花青素的摩爾比為1.1:1。
(2)帶保護基團的多肽修飾的三聚原花青素的合成
①配製第三種溶液
在常壓、室溫條件下將步驟(1)製備的27.5mg丙烯醯基修飾的三聚原花青素溶解於10mL去離子水中形成第三種溶液;
②配製第四種溶液
以40mg結構式如式(Ⅲ)所示的多肽為溶質,在室溫、常壓條件下將所述溶質溶解於10mL去離子水中形成第四種溶液;
③合成
將二甲基苯基磷加入第三種溶液中,在室溫、常壓攪拌混合均勻,然後加入第四種溶液並攪拌混合均勻,在密封條件下於室溫、常壓反應24h,然後在去離子水中用截留分子量為1500道爾頓的透析袋透析1天,冷凍乾燥,即得帶保護基團的多肽修飾的三聚原花青素;
所述第四種溶液的加入量應使第四種溶液中的巰基與第三種溶液中的碳碳雙鍵的摩爾比為1.1:1,所述二甲基苯基磷與第四種溶液中的巰基的摩爾比為5×10-6:1。
(3)多肽修飾的三聚原花青素的合成
①配製第五種溶液
在室溫、常壓條件下將氨水、二氧六環、濃度為5mol/L的氫氧化鈉水溶液混合均勻得到第五種溶液,第五種溶液中,氨水、二氧六環、氫氧化鈉的摩爾比為2:0.9:0.1;
②合成
以40mg步驟(2)製備的帶保護基團的多肽修飾的三聚原花青素為溶質,在室溫、常壓條件下將所述溶質溶解於2mL第五種溶液中並攪拌混合均勻,在密封條件下於室溫、常壓反應24h,然後然後在去離子水中用截留分子量為1000道爾頓的透析袋透析24h,冷凍乾燥,即得多肽修飾的三聚原花青素,記作SAP-3-OPC。
實施例3
本實施例中,提供多肽修飾的四聚原花青素的製備方法,其合成路線如圖1所示,步驟如下:
(1)丙烯醯基修飾的四聚原花青素的合成
①配製第一種溶液
在室溫、常壓條件下將1.18g四聚原花青素溶解於40mL二甲基甲醯胺中形成第一種溶液,四聚原花青素在第一種溶液中的濃度為29.5mg/mL;
②配製第二種溶液
在室溫、常壓條件下將三乙胺溶解於第一種溶液中形成第二種溶液,所述三乙胺與第一種溶液中四聚原花青素的摩爾比為1:1;
③合成
在0~5℃的冰水浴及氬氣保護條件下將丙烯醯氯滴加到第二種溶液中,在室溫室溫、常壓條件下反應30h,然後在去離子水中用截留分子量為750道爾頓的透析袋透析1天,再冷凍乾燥,即得丙烯醯基修飾的四聚原花青素;所述丙烯醯氯與第二種溶液中四聚原花青素的摩爾比為1:1。
(2)帶保護基團的多肽修飾的四聚原花青素的合成
①配製第三種溶液
在常壓、室溫條件下將步驟(1)製備的36.6mg丙烯醯基修飾的四聚原花青素溶解於10mL去離子水中形成第三種溶液;
②配製第四種溶液
以40mg結構式如式(Ⅲ)所示的多肽為溶質,在室溫、常壓條件下將所述溶質溶解於10mL去離子水中形成第四種溶液;
③合成
將二甲基苯基磷加入第三種溶液中,在室溫、常壓攪拌混合均勻,然後加入第四種溶液並攪拌混合均勻,在密封條件下於室溫、常壓反應30h,然後在去離子水中用截留分子量為2000道爾頓的透析袋透析1天,冷凍乾燥,即得帶保護基團的多肽修飾的四聚原花青素;
所述第四種溶液的加入量應使第四種溶液中的巰基與第三種溶液中的碳碳雙鍵的摩爾比為1:1,所述二甲基苯基磷與第四種溶液中的巰基的摩爾比為8×10-6:1。
(3)多肽修飾的四聚原花青素的合成
①配製第五種溶液
在室溫、常壓條件下將氨水、二氧六環、濃度為3mol/L的氫氧化鈉水溶液混合均勻得到第五種溶液,第五種溶液中,氨水、二氧六環、氫氧化鈉的摩爾比為2:1:0.1;
②合成
以60mg步驟(2)製備的帶保護基團的多肽修飾的四聚原花青素為溶質,在室溫、常壓條件下將所述溶質溶解於3mL第五種溶液中並攪拌混合均勻,在密封條件下於室溫、常壓反應30h,然後然後在去離子水中用截留分子量為1500道爾頓的透析袋透析30h,冷凍乾燥,即得多肽修飾的四聚原花青素,記作SAP-4-OPC。
實施例4
本實施例中,提供多肽修飾的五聚原花青素的製備方法,其合成路線如圖1所示,步驟如下:
(1)丙烯醯基修飾的五聚原花青素的合成
①配製第一種溶液
在室溫、常壓條件下將1.475g五聚原花青素溶解於40mL二甲基甲醯胺中形成第一種溶液,五聚原花青素在第一種溶液中的濃度為36.9mg/mL;
②配製第二種溶液
在室溫、常壓條件下將三乙胺溶解於第一種溶液中形成第二種溶液,所述三乙胺與第一種溶液中五聚原花青素的摩爾比為1.5:1;
③合成
在-5~0℃及氬氣保護條件下將丙烯醯氯滴加到第二種溶液中,在室溫室溫、常壓條件下反應32h,然後在去離子水中用截留分子量為1000道爾頓的透析袋透析1天,再冷凍乾燥,即得丙烯醯基修飾的五聚原花青素;所述丙烯醯氯與第二種溶液中五聚原花青素的摩爾比為1.5:1。
(2)帶保護基團的多肽修飾的五聚原花青素的合成
①配製第三種溶液
在常壓、室溫條件下將步驟(1)製備的45.8mg丙烯醯基修飾的五聚原花青素溶解於10mL去離子水中形成第三種溶液;
②配製第四種溶液
以40mg結構式如式(Ⅲ)所示的多肽為溶質,在室溫、常壓條件下將所述溶質溶解於10mL去離子水中形成第四種溶液;
③合成
將二甲基苯基磷加入第三種溶液中,在室溫、常壓攪拌混合均勻,然後加入第四種溶液並攪拌混合均勻,在密封條件下於室溫、常壓反應32h,然後在去離子水中用截留分子量為2500道爾頓的透析袋透析1天,冷凍乾燥,即得帶保護基團的多肽修飾的五聚原花青素;
所述第四種溶液的加入量應使第四種溶液中的巰基與第三種溶液中的碳碳雙鍵的摩爾比為1.5:1,所述二甲基苯基磷與第四種溶液中的巰基的摩爾比為10-5:1。
(3)多肽修飾的五聚原花青素的合成
①配製第五種溶液
在室溫、常壓條件下將氨水、二氧六環、濃度為4mol/L的氫氧化鈉水溶液混合均勻得到第五種溶液,第五種溶液中,氨水、二氧六環、氫氧化鈉的摩爾比為2:0.95:0.1;
②合成
以80mg步驟(2)製備的帶保護基團的多肽修飾的五聚原花青素為溶質,在室溫、常壓條件下將所述溶質溶解於4mL第五種溶液中並攪拌混合均勻,在密封條件下於室溫、常壓反應32h,然後然後在去離子水中用截留分子量為2000道爾頓的透析袋透析30h,冷凍乾燥,即得多肽修飾的五聚原花青素,記作SAP-5-OPC。
實施例5
本實施例中,測定實施例1製備的SAP-2-OPC在牙釉質表面的吸附情況,具體使用雷射共聚焦顯微鏡觀察螢光標記了的SAP-2-OPC和二聚原花青素在牙釉質表面的吸附情況。
(1)螢光標記
螢光標記物質選擇羅丹明,標記方法為:分別向SAP-2-OPC和二聚原花青素的二甲基亞碸溶液中加入與SAP-2-OPC、二聚原花青素等摩爾的羅丹明的二甲基亞碸溶液,再分別加入與SAP-2-OPC、二聚原花青素等摩爾的1-(3-二甲氨基丙基)-3-乙基碳二亞胺(EDC),攪拌1天,得到濃度為2.25mg/mL的羅丹明標記的SAP-2-OPC和濃度為2.25mg/mL羅丹明標記的二聚原花青素。
(2)酸蝕和未酸蝕牙釉質塊的製備
①取因正畸拔除的正常前磨牙,用乙醇除去正常牙表面的有機汙染物,用蒸餾水衝洗乾淨後貯存於4℃的濃度為0.05wt%的麝香草酚水溶液中備用。
②使用高速牙塊切割機上將需要的牙釉質部分從經過步驟①處理的牙上切下,切割成4×4×2mm大小的牙齒塊,切割牙釉質面暴露,其餘部分用甲基丙烯酸甲酯樹脂包埋,將暴露面用依次用800#、1200#、2400#碳化矽砂紙在流水下磨平、拋光,去除表層約150μm,以消除表面有機汙染物和表面的不規則,得到未酸蝕牙釉質塊,置於濃度為0.05wt%的麝香草酚水溶液中備用,使用前,將樹脂包埋的牙塊置於去離子水中,用超聲波清洗器中處理30min。
③使用高速牙塊切割機上將需要的牙釉質部分從經過步驟①處理的牙上切下,切割成4×4×2mm大小的牙齒塊,切割牙釉質面暴露,其餘部分用甲基丙烯酸甲酯樹脂包埋,將暴露面用依次用800#、1200#、2400#碳化矽砂紙在流水下磨平、拋光,去除表層約150μm,以消除表面有機汙染物和表面的不規則,得到未酸蝕牙釉質塊,置於濃度為0.05wt%的麝香草酚水溶液中備用,使用前,將樹脂包埋的牙塊置於去離子水中,用超聲波清洗器中處理30min,取出放入37wt%的磷酸中浸泡45s(每顆樹脂包埋的牙塊用10mL磷酸浸泡),用PBS衝洗3次,再將其置於去離子水中,用超聲波清洗器中處理5min,將得到的酸蝕牙釉質塊保存於磷酸鹽緩衝液(PBS)中待用。
(3)用移液槍分別量取2mL羅丹明標記的SAP-2-OPC溶液浸泡酸蝕和未酸蝕牙釉質塊,用移液槍分別量取2mL羅丹明標記的二聚原花青素溶液浸泡未酸蝕牙釉質塊,浸泡1天後用PBS衝洗3次,將樣品烘乾後用雷射共聚焦顯微鏡觀察其表面的螢光分布情況,結果如圖6所示。
圖6中,圖(A)~(C)分別為羅丹明標記的SAP-2-OPC在酸蝕牙釉質塊上的螢光分布圖、羅丹明標記的SAP-2-OPC在未酸蝕牙釉質塊上的螢光分布圖、羅丹明標記的二聚原花青素在未酸蝕牙釉質塊上的螢光分布圖,其中,圖(A)中樣品具有明顯的紅色螢光,圖(B)中樣品的螢光較圖(A)更暗,這是由於酸蝕後的牙釉質的比表面積增大,能吸附更多的螢光標記的SAP-2-OPC造成的,而圖(C)中樣品沒有螢光,說明二聚原花青素對牙釉質沒有吸附作用。本實施例說明本發明所述多肽修飾的低聚原花青素在牙釉質上具有良好的吸附性能。
實施例6
本實施例中,測定實施例1製備的SAP-2-OPC和二聚原花青素在羥基磷灰石粉末上的吸附情況,所述羥基磷灰石粉末的規格為HA/M30,其含水量低於0.1%。
將SAP-2-OPC溶解在去離子水中,配製成SAP-2-OPC濃度分別為0.25mg/mL、0.75mg/mL、1.25mg/mL、1.75mg/mL、2.25mg/mL及2.75mg/mL SAP-2-OPC水溶液,用紫外可見分光光度計測量前述6個濃度的SAP-2-OPC水溶液在波長為264nm處的吸光度值,得到SAP-2-OPC濃度與吸光度值之間的線性關係,即標準曲線。
將SAP-2-OPC溶解在去離子水中,配製成SAP-2-OPC濃度分別為0.25mg/mL、0.75mg/mL、1.25mg/mL、1.75mg/mL、2.25mg/mL及2.75mg/mL的SAP-2-OPC水溶液,分別記作1~6#溶液。取量程為10mL的離心PE管6支,分別向其中加入50mg羥基磷灰石粉末,然後取1~6#溶液各1mL加入離心PE管中,在37℃振蕩攪拌12h,然後對各離心PE管中的混合液以10000轉/分鐘的轉速離心4分鐘,取離心所得上清液用紫外分光光度計測量上清液中SAP-2-OPC的濃度,對照標準曲線,計算出吸附在羥基磷灰石粉末上的SAP-2-OPC的量,結果如圖7所示。由圖7可知,當SAP-2-OPC的濃度為2.25mg/mL時在羥基磷灰石粉末上達吸附飽和,SAP-2-OPC在50mg羥基磷灰石粉末上的飽和吸附量為1.9mg。
二聚原花青素在羥基磷灰石粉末上的吸附情況的測試方法與上述SAP-2-OPC在羥基磷灰石粉末上的吸附情況的測試方法類似,測試結果表明二聚原花青素在羥基磷灰石上的飽和吸附量為0,即二聚原花青素對羥基磷灰石粉末並沒有吸附作用。
實施例7
本實施例中,測定實施例1製備的SAP-2-OPC在羥基磷灰石片上聚集成膜的情況,所述羥基磷灰石片的尺寸為8mm*2mm,其含水量低於0.1%。
採用掃描電鏡觀察SAP-2-OPC在羥基磷灰石片表面聚集成膜的情況。將SAP-2-OPC溶解在去離子水中,配製成SAP-2-OPC濃度為2.25mg/mL的SAP-2-OPC水溶液,用移液槍量取2mL的SAP-2-OPC水溶液浸泡羥基磷灰石片,浸泡1天後用PBS衝洗3次,烘乾後用掃描觀察SAP-2-OPC在羥基磷灰石片表面聚集成膜的情況,結果如圖8所示,其中,圖(A)~(C)是SAP-2-OPC在羥基磷灰石片表面聚集成膜後不同放大倍數的SEM照片。由圖8可知,本發明所述多肽修飾的低聚原花青素在羥基磷灰石片上具有良好的聚集成膜性能。
實施例8
本實施例中,測定實施例1製備的SAP-2-OPC對牙釉質再礦化的促進能力。
(1)酸蝕牙釉質塊的製備
①取因正畸拔除的正常前磨牙,用乙醇除去正常牙表面的有機汙染物,用蒸餾水衝洗乾淨後貯存於4℃的濃度為0.05wt%的麝香草酚水溶液中備用。
②使用高速牙塊切割機上將需要的牙釉質部分從經過步驟①處理的牙上切下,切割成4×4×2mm大小的牙齒塊,切割牙釉質面暴露,其餘部分用甲基丙烯酸甲酯樹脂包埋,將暴露面用依次用800#、1200#、2400#碳化矽砂紙在流水下磨平、拋光,去除表層約150μm,以消除表面有機汙染物和表面的不規則,得到未酸蝕牙釉質塊,置於濃度為0.05wt%的麝香草酚水溶液中備用,使用前,將樹脂包埋的牙塊置於去離子水中,用超聲波清洗器中處理30min,取出放入37wt%的磷酸中浸泡45s(每顆樹脂包埋的牙塊用10mL磷酸浸泡),用PBS衝洗3次,再將其置於去離子水中,用超聲波清洗器中處理5min,將得到的酸蝕牙釉質塊保存於磷PBS中待用。
(2)①用去離子水將實施例1製備的SAP-2-OPC配製成濃度為2.25mg/mL的SAP-2-OPC水溶液。
②按如下配方配製模擬唾液:CaCl2 0.1665g、KCl 9.685g、NaH2PO40.1224g、NaN3·0.065g、HEPES 4.766g,在37℃溶於用750mL去離子中,用1mol/L的KOH溶液調節pH至7.02,用去離子水定容至1000mL。
(3)將步驟(1)製備的酸蝕牙釉質塊分為實驗組和空白組,每組中酸蝕牙釉質塊2塊。各組的操作如下:
①將實驗組的酸蝕牙釉質塊在室溫下浸泡於2.25mg/mL的SAP-2-OPC水溶液中,浸泡時間為30min,取出,用PBS洗滌;將空白組的酸蝕牙釉質塊在室溫下浸泡於去離子水中,浸泡時間為30min,取出,用PBS洗滌。
②將步驟①得到的牙釉質塊浸泡於模擬唾液中,於37℃恆溫保存,模擬唾液每1天更換一次,並重複步驟①,每組中的酸蝕牙釉質塊分別浸泡1天和2周,取出,用PBS洗滌、烘乾,然後用掃描電鏡觀察各組酸蝕牙釉質塊表面羥基磷灰石的沉積和結晶的情況,結果如圖9和圖10所示。
圖9中,圖(A)、(B)分別為實驗組礦化1天和2周後酸蝕牙釉質塊表面的SEM圖,圖10中,圖(A)、(B)分別為空白組礦化1天和2周後酸蝕牙釉質塊表面的SEM圖。由圖9可知,實驗組的酸蝕牙釉質塊在模擬唾液中浸泡1天後,其表面已有羥基磷灰石的沉積和結晶,實驗組的酸蝕牙釉質在模擬唾液中浸泡2周後,其表面明顯可見有一定厚度的羥基磷灰石的沉積和結晶,而由圖10可知,空白組的酸蝕牙釉質塊則礦化程度較弱,說明本發明所述多肽修飾的低聚原花青素在模擬唾液中能誘導羥基磷灰石在酸蝕牙釉質塊表面沉積和結晶,可用作牙釉質原位再礦化誘導劑。
實施例9
本實施例中,考察三價鐵離子對本發明所述多肽修飾的低聚原花青素對牙釉質再礦化能力的促進作用。
(1)酸蝕牙釉質塊的製備
①取因正畸拔除的正常前磨牙,用乙醇除去正常牙表面的有機汙染物,用蒸餾水衝洗乾淨後貯存於4℃的濃度為0.05wt%的麝香草酚水溶液中備用。
②使用高速牙塊切割機上將需要的牙釉質部分從經過步驟①處理的牙上切下,放置於規格為1×1×1cm的砂盤中,並向沙盤中澆上DMDHE樹脂,待其自然固化,將樹脂塊取出,在高速渦輪砂紙上將其磨平,特別注意將所需的牙釉質表面從樹脂中暴露出來。將包有DMDHE樹脂的牙塊置於去離子水中,在超聲波清洗器中處理30min,取出放入37wt%的磷酸中浸泡45s(每顆樹脂包埋的牙塊用10mL磷酸浸泡),用PBS衝洗3次,再將其置於去離子水中,在超聲波清洗器中處理5min,將得到的酸蝕牙釉質塊保存於PBS中待用。
(2)①用去離子水將實施例1製備的SAP-2-OPC配製成濃度為2.25mg/mL的SAP-2-OPC水溶液。
②按如下配方配製模擬唾液:CaCl2 0.1665g、KCl 9.685g、NaH2PO40.1224g、NaN3·0.065g、HEPES 4.766g,在37℃溶於用750mL去離子中,用1mol/L的KOH溶液調節pH至7.02,用去離子水定容至1000mL。
(3)配製Fe3+緩衝液
①配製Tris.HCl緩衝液,配方為:50mL 0.1mol/L Tris水溶液、45.7mL 0.1mol/L HCl水溶液用去離子水定容至100mL,用0.1mol/lNaOH調節pH值至7;
②用Tris.HCl緩衝液將實施例1製備的SAP-2-OPC配製成SAP-2-OPC濃度為2.25mg/mL的SAP-2-OPC溶液,用Tris.HCl緩衝液將FeCl3.6H2O配製成Fe3+與前述SAP-2-OPC溶液中SAP-2-OPC摩爾濃度相等的溶液,然後將兩種溶液等摩爾比混合,並用0.1mol/L的NaOH調pH值至9得到Fe3+緩衝液。
(4)將步驟(1)製備的酸蝕牙釉質塊分為實驗組和空白組,每組中酸蝕牙釉質塊2塊。各組的操作如下:
①將實驗組的酸蝕牙釉質塊在室溫下浸泡於步驟(3)配製的Fe3+緩衝液中,浸泡時間為30min,取出,用PBS洗滌;將空白組的酸蝕牙釉質塊在室溫下浸泡於去離子水中,浸泡時間為30min,取出,用PBS洗滌。
②將步驟①中得到的牙釉質塊浸泡於模擬唾液中,於37℃恆溫保存,模擬唾液每1天更換一次,並重複步驟①,分別浸泡1天、2周,取出,用PBS洗滌、烘乾,然後用掃描電鏡觀察各組酸蝕牙釉質塊表面羥基磷灰石的沉積和結晶的情況,結果如圖10和圖11所示。
圖11中,圖(A)、(B)分別為實驗組的酸蝕牙釉質塊在礦化1天和2周後,其表面的SEM圖,由圖11可知,酸蝕牙釉質塊表面明顯可見有一定厚度的羥基磷灰石的沉積和結晶,與實例8中實驗組的酸蝕牙釉質塊(圖9)相比對比,礦化程度明顯增強並且分布更加均勻,說明三價鐵離子能促進本發明所述多肽修飾的低聚原花青素誘導羥基磷灰石在酸蝕牙釉質塊表面沉積和結晶的能力,三價鐵離子可與本發明所述多肽修飾的低聚原花青素配合作為牙釉質原位再礦化誘導劑應用。
實施例5~9說明本發明所述多肽修飾的低聚原花青素可作為牙釉質原位再礦化誘導劑應用。
實施例10
本實施例中,考察實施例1製備的SAP-2-OPC對口腔變異鏈球菌的抑制作用。
(1)將SAP-2-OPC溶解在去離子水中,配製成SAP-2-OPC濃度為2.25mg/mL的SAP-2-OPC水溶液,用移液槍量取2mL的SAP-2-OPC水溶液浸泡羥基磷灰石片1天,乾燥後,得到帶SAP-2-OPC塗層的在羥基磷灰石片。
(2)以步驟(1)製備的帶SAP-2-OPC塗層的羥基磷灰石片作為實驗組試樣,以羥基磷灰石片作為空白組試樣,實驗組和空白組的操作如下:
實驗組:將帶SAP-2-OPC塗層的羥基磷灰石片置於孔板中,將口腔變異鏈球菌接種到所述帶SAP-2-OPC塗層的羥基磷灰石片表面,在厭氧培養箱中培養24h,然後將所述帶SAP-2-OPC塗層的羥基磷灰石片移到新孔板,用PBS衝洗3次,將濃度為5mg/mL的MTT加入到放有所述帶SAP-2-OPC塗層的羥基磷灰石片的孔板中,在厭氧培養箱中培養1h,將孔板中的MTT吸盡,加DMSO,放入搖床振搖至所述帶SAP-2-OPC塗層的羥基磷灰石上的甲瓚完全溶解,吸取所得溶液到96孔板中,用酶標儀在波長600nm下測吸光度值OD600。
空白組:將羥基磷灰石片置於孔板中,將口腔變異鏈球菌接種到羥基磷灰石片表面,在厭氧培養箱中培養24h,然後將羥基磷灰石片移到新孔板,用PBS衝洗3次,將濃度為5mg/mL的MTT加入到放有羥基磷灰石片的孔板中,在厭氧培養箱中培養1h,將孔板中的MTT吸盡,加DMSO,放入搖床振搖至所述帶SAP-2-OPC塗層的羥基磷灰石上的甲瓚完全溶解,吸取所得溶液到96孔板中,用酶標儀在波長600nm下測吸光度值OD600。
實驗組和空白組的實驗結果如圖12所示,由圖12可知,空白組的吸光度值明顯大於實驗組,說明SAP-2-OPC具有抑菌作用。
(3)以步驟(1)製備的帶SAP-2-OPC塗層的羥基磷灰石片作為實驗組試樣,以羥基磷灰石片作為空白組試樣,實驗組和空白組的操作如下:
分別將口腔變異鏈球菌接種到帶SAP-2-OPC塗層的羥基磷灰石片和羥基磷灰石片表面,在厭氧培養箱中培養24小時,用戊二醛固定過夜,然後分別依次用體積濃度分別為60%、70%、80%、90%的乙醇脫水10分鐘。用掃描電鏡觀察,結果如圖13所示,其中,圖(A)、(B)分別為實驗組的帶SAP-2-OPC塗層的羥基磷灰石片表面的SEM照片,圖(C)、(D)分別為空白組的羥基磷灰石片表面的SEM照片,由圖13可知,與空白組相比,帶SAP-2-OPC塗層的羥基磷灰石片表面粘附的變異鏈球菌數量明顯減少。
(4)以步驟(1)製備的帶SAP-2-OPC塗層的羥基磷灰石片作為實驗組試樣,以羥基磷灰石片作為空白組試樣,實驗組和空白組的操作如下:
分別將口腔變異鏈球菌接種到帶SAP-2-OPC塗層的羥基磷灰石片和羥基磷灰石片表面,在厭氧培養箱中培養24小時,然後分別將細胞死活染染料滴到經過上述處理的帶SAP-2-OPC塗層的羥基磷灰石片和羥基磷灰石片上,染色15分鐘,用共聚焦顯微鏡觀察,結果如圖14所示。
圖14中,圖(A)、(B)分別為空白組的羥基磷灰石片表面的活菌和死菌被染色後的共聚焦顯微鏡圖,圖(C)、(D)分別為實驗組的帶SAP-2-OPC塗層的羥基磷灰石片表面的活菌和死菌被染色後的共聚焦顯微鏡圖,將圖(A)與(B),(C)與(D)比較可知,活菌和死菌的位置基本一致,說明活菌和死菌都會粘附在羥基磷灰石片上,將圖(A)與圖(C)、圖(B)與圖(D)比較可知,實驗組的帶SAP-2-OPC塗層的羥基磷灰石片上比空白組的羥基磷灰石片上粘附的活細菌和死細菌數量都明顯減少,說明SAP-2-OPC具有良好的抑菌作用是由於其能有效防止細菌粘附,進而阻止粘附的細菌在其上繁殖。本實施例說明本發明所述多肽修飾的低聚原花青素可作為牙科植入體表面抗菌材料應用。