一種2‑位噻吩基取代的非對稱型氟硼絡合二吡咯甲川衍生物及其製備方法與流程
2023-05-13 16:52:21 3
本發明涉及一種2-位噻吩基取代的非對稱型氟硼絡合二吡咯甲川衍生物及其製備方法,該類衍生物可應用於螢光染料、發光材料、光伏材料、生命科學、分析科學、環境能源科學、有機太陽能電池等領域。
背景技術:
氟硼絡合二吡咯甲川(BODIPY)衍生物是一類良好的光敏染料,具有良好的穩定性,高的摩爾消光係數 (1×105 M-1 cm-1) 和較高的氧化電位,並且其可以修飾的位置多,可通過選擇性的接入具有相應功能的基團來調節BODIPY染料的吸收/發射波長等光學性質、穩定性、以及溶解性等物理性質。自1968年被首次報導以後就受到人們的關注,近二十年來更是被廣泛研究,已被運用到螢光染料、發光材料、光伏材料、生命科學、分析科學、環境科學、能源科學、有機太陽能電池等領域。
在小分子太陽能電池給體材料中,分子一般都是對稱的結構,非對稱的D-A分子還很少有文獻報導。為了改良一些對稱的D-A分子的光譜吸收性質,本發明設計了一種以BODIPY為中心吸電子基元,2,6-位分別引入噻吩和芴、咔唑以及三苯胺等基團,構建了一系列結構新穎的非對稱BODIPY衍生物,並研究它們的光物理性質、電化學行為,以揭示這類分子結構與性質之間的關係。
研究結果表明,這類分子的吸收光譜寬闊,摩爾消光係數高,分子內電荷遷移和能量轉移均較為順暢,適合構建一些有機光電子材料,如有機太陽能電池材料等。
技術實現要素:
本發明的目的是提供一種2-位噻吩基取代的非對稱D-A型氟硼絡合二吡咯甲川(BODIPY)衍生物,其具有較為穩定、寬闊、強烈的光譜吸收,材料在空氣中穩定性好。
本發明的另一個目的是提供一種2-位噻吩基取代的非對稱型氟硼絡合二吡咯甲川衍生物的製備方法,其製備成本低、反應條件溫和、易於控制。
為實現上述目的,本發明採用以下技術方案:一種2-位噻吩基取代的非對稱型氟硼絡合二吡咯甲川衍生物,具有通式Ⅰ的化學結構:
Ⅰ
式中,
n為1-20的自然數。
一種2-位噻吩基取代的非對稱型氟硼絡合二吡咯甲川衍生物的製備方法,包括以下步驟:
(1) 2-碘-9,9-二甲基芴與雙聯頻哪醇硼酯反應,得到中間體1,其結構為:
(2) 3-溴-9-辛基咔唑與雙聯頻哪醇硼酯反應,得到中間體2,其結構為:
(3) 4-溴-N,N-二{4-[2-(2-甲基庚基)]苯基}苯胺與雙聯頻哪醇硼酯反應,得到中間體3,其結構為:
(4)對羥基苯甲醛與溴代烷烴反應,然後再與新蒸吡咯和三氟化硼-乙醚反應,最後再經N-溴代丁二醯亞胺溴化,得到中間體4,其結構為:
(5) 中間體4和2-(三丁基錫)噻吩通過Stille偶聯得到中間體5,其結構為:
(6) 中間體1和中間體5在催化劑的催化下反應生成目標產物BDP1,其結構為:
(7) 中間體2和中間體5 在催化劑的催化下反應生成目標產物BDP2,其結構為:
(8) 中間體3和中間體5在催化劑的催化下反應生成目標產物BDP3,其結構為:
作為上述技術方案的優選,步驟(1)-(8)中所述反應的反應介質為正己烷、四氫呋喃、N,N-二甲基甲醯胺、二氯甲烷、甲苯、氯仿、石油醚、乙酸乙酯、乙醇的一種或幾種混合。
作為上述技術方案的優選,步驟(4)、(5)、(6)、(7)、(8)中所述催化劑為四(三苯基膦)鈀、雙(三苯基膦)二氯化鈀、三(二亞苄基丙酮)二鈀、[1,1'-雙(二苯基磷)二茂鐵]二氯化鈀、 三氯化銦中的一種或幾種混合。
作為上述技術方案的優選,步驟(5)中,合成中間體5的反應中,中間體4和2-(三丁基錫)噻吩的摩爾比為1:1.1-1:2。
作為上述技術方案的優選,步驟(5)中,所述反應的反應溫度為90-150℃。
作為上述技術方案的優選,步驟(6)、(7)、(8)中,所述反應的反應溫度為60-120℃。
作為上述技術方案的優選,步驟(5)-(8)中,所述反應的反應時間為12-36h。
與現有技術相比,本發明具有以下有益效果:
(1)本發明通過一系列反應合成中間體5,最後再利用該中間體與多種修飾基團進行Suzuki/Stille偶聯反應,得到2-位噻吩基取代,6-位分別為芴、咔唑和三苯胺等強給電子單元取代的目標染料分子。
(2)本發明提供的合成方法製得的目標產物合成成本低、反應溫和、易於控制。
(3)通過對幾種目標產物的光譜和電化學數據分析,可以看出這些分子的吸收光譜寬闊,摩爾消光係數高。電化學測試結果顯示:幾種目標產物的HOMO能級分別在-5.2 ~ -5.4 eV之間,LUMO能級在-3.3 ~ -3.4 eV。
附圖說明:
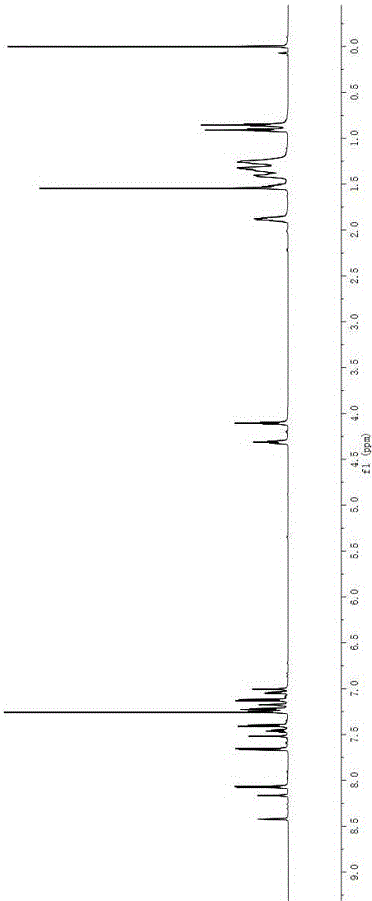
圖1為本發明實施例1的BDP11HNMR圖。
圖2為本發明實施例1的BDP21HNMR圖。
圖3為本發明實施例1的BDP31HNMR圖。
圖4為本發明實施例1的BDP113C NMR圖。
圖5為本發明實施例1的BDP213C NMR圖。
圖6為本發明實施例1的 BDP1質譜圖。
圖7為本發明實施例1的BDP2質譜圖。
圖8為本發明實施例1的BDP3質譜圖。
具體實施方式:
為更好的理解本發明,下面通過實施例及附圖對本發明進一步說明,實施例只用於解釋本發明,不會對本發明構成任何的限定。
實施例1
中間體1的合成:
在2-碘-9,9』-二甲基芴 (3.2 g, 10 mmol)、雙聯頻哪醇硼酯 (2.79 g, 11 mmol)、乙酸鉀 (2.94 g, 30 mmol)、DMF (80 mL)的混合液中加入Pd(dppf)Cl2 (360 mg, 0.5 mmol)。體系在氬氣保護下, 90oC反應8 h。停止反應,混合物冷卻至室溫後倒入200 mL 水中,用乙酸乙酯萃取 (50 mL × 3),再用飽和食鹽水洗滌數次,合併有機相,無水硫酸鎂乾燥。減壓旋轉蒸發,去除溶劑,殘餘物以石油醚:乙酸乙酯 = 20:1 (v/v) 為洗脫劑用矽膠 (200-300目) 進行柱層析分離,得到白色固體2.82 g,產率88%。1H NMR (600 MHz, CDCl3) δ: 7.91 (s, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.76 (t, J = 6.8 Hz, 2H), 7.48-7.45 (m, 1H), 7.37-7.34 (m, 2H), 1.53 (s, 6H), 1.40 (s, 12H)。
中間體2的合成:
在3-溴-9-辛基咔唑 (2 g, 5.6 mmol)、雙聯頻哪醇硼酯 (1.7 g, 6.7 mmol)、乙酸鉀 (2.7 g, 28 mmol)、DMF (80 mL)的混合液中加入Pd(dppf)Cl2 (150 mg, 0.21 mmol)。體系在氬氣保護下, 90oC反應8 h。停止反應,混合物冷卻至室溫後倒入200 mL 水中,用二氯甲烷萃取 (50 mL × 3),再用飽和食鹽水洗滌數次,合併有機相,無水硫酸鎂乾燥。減壓旋轉蒸發,去除溶劑,殘餘物以石油醚:乙酸乙酯 = 20:1 (v/v) 為洗脫劑用矽膠 (200-300目) 進行柱層析分離,得到白色固體1.7 g,產率76%。1H NMR (600 MHz, CDCl3), δ: 8.60 (s, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.45 (d, J = 7.3 Hz, 1H), 7.40 (t, J = 9.5 Hz, 2H), 7.23 (d, J = 7.0 Hz, 1H), 4.30 (t, J = 6.9 Hz, 2H), 1.90-1.86 (m, 2H), 1.44 (s, 12H), 1.36-1.28 (m, 10H), 0.85 (t, J = 13.6 Hz, 3H). MALDI-TOF-MS, m/z: calcd for C26H36BNO2 [M]+: 405.300; found 405.333。
中間體3的合成:
中間體3 的反應條件與合成中間體2的條件類似,用4-溴-N,N-二(4-異辛基苯基)苯胺作為反應底物。純化後得到白色固體, 產率84%。1H NMR (600 MHz, CDCl3), δ: 7.64 (d, J = 8.5 Hz, 2H), 7.25 (d, J = 8.6 Hz, 4H), 7.01 (d, J = 8.6 Hz, 4H), 6.98 (d, J = 8.5 Hz, 2H), 1.71 (s, 4H), 1.42-1.35 (m, 12H), 1.33 (s, 12H), 0.78-0.71 (m, 18H)。
中間體4的合成:
在100 mL單口瓶中加入對辛氧基苯甲醛 (2.34 g, 10 mmol) 和新蒸的吡咯 (30 mL, 430 mmol), 向溶液中鼓氬氣置換10 min,在氬氣保護下迅速加入催化劑InCl3 (0.11 g, 0.5 mmol), 在室溫下磁力攪拌5 h, 向其中加入NaOH (0.2 g, 5 mmol)粉末繼續攪拌30 min,終止反應。減壓蒸餾,回收多餘的吡咯,得粗產品,用矽膠(200-300目)柱層析[洗脫液,V(石油醚):V(乙酸乙酯) = 7:1],純化得到白色晶體2.45 g, 產率70%。
在500 mL三口瓶中,取上述合成的白色晶體 (3.50 g, 10 mmol) 溶解於二氯甲烷 (80 mL) 中,加入四氯苯醌 (2.9 g, 12 mmol), 室溫條件下磁力攪拌,充分氧化8 h。然後將反應混合液置於氬氣保護下,慢慢滴加三氟化硼乙醚 (37 mL, 300 mmol),攪拌反應10 min 後再緩慢加入三乙胺 (41.7 mL, 300 mmol),滴加完畢後繼續反應8 h。混合物倒入氫氧化鈉溶液中,用二氯甲烷萃取。有機相用無水硫酸鈉乾燥。過濾、濾液減壓下去除溶劑。粗產品用矽膠 (200-300目) 柱層析[洗脫液,V(石油醚):V(乙酸乙酯)=10:1] 分離得紅綠色粉末狀化合物2.50 g,產率63%。
向100 mL三口瓶中依次加入上一步合成的紅綠色固體 (1.0 g, 2.5 mmol)、DMF (20 mL) 和二氯甲烷 (20 mL),在室溫下磁力攪拌幾分鐘後,裝上恆壓滴液漏鬥,抽真空,通入氬氣保護,將NBS (0.89 g, 5.0 mmol) 溶解在DMF (5 mL) 和二氯甲烷 (5 mL)中,通過恆壓滴液漏鬥逐滴滴入到三口瓶中,滴加完畢後再繼續反應4 h,整個過程避光。向三口瓶中加入50 mL去離子水,用二氯甲烷萃取三次 (80 mL × 3),有機層再經飽和食鹽水洗滌 (100 mL × 3),合併有機相,無水硫酸鎂乾燥過夜。過濾收集濾液,減壓下旋轉蒸發去除溶劑,得粗產品,用矽膠(200-300目)柱層析[洗脫液,V(石油醚):V(乙酸乙酯) = 20:1]純化得紅色粉末狀固體 0.97 g,產率70%。1H NMR (600 MHz, CDCl3), δ: 7.84 (d, J = 6.4 Hz, 2H), 7.53 (d, J = 8.6 Hz, 2H), 7.07 (d, J = 8.7 Hz, 2H), 7.02 (s, 2H), 4.09 (t, J = 6.5 Hz, 2H), 1.92–1.82 (m, 2H), 1.52 (m, 10H), 0.91 (t, 3H). 13C NMR (151 MHz, CDCl3), δ: 162.72, 147.50, 143.22, 134.70, 132.93, 131.16, 125.48, 114.46, 106.99, 68.25, 30.91, 29.70, 29.31, 28.78, 25.61, 22.42, 13.93. MALDI-TOF-MS, m/z: calcd for C23H25BBr2F2N2O [M-47]+: 554.000; found 507.100。
中間體5的合成:
在100 mL單口瓶中,依次加入中間體4 (0.55 g, 1.0 mmol)、2-(三丁基錫)噻吩(0.45 g, 1.2 mmol), 氟化銫 (0.3 g, 2.0 mmol) 和甲苯 (30 mL),抽真空,再向反應瓶中加入四(三苯基膦)鈀 (0.15 g, 0.13 mmol),氬氣保護,110oC下磁力攪拌反應24 h。停止反應,冷卻至室溫,倒入水中,用乙酸乙酯萃取,再用飽和食鹽水洗滌 (30 mL × 3),收集有機相,無水硫酸鈉乾燥。減壓下旋轉蒸發去除溶劑,得粗產品,用矽膠 (300-400目) 柱層析 [洗脫液,V(石油醚):V(乙酸乙酯) = 10:1],得到褐色粉末狀化合物固體0.31 g,產率56%。1H NMR (400 MHz, CDCl3), δ: 8.25 (s, 1H), 7.82 (s, 1H), 7.60 (d, J = 3.6 Hz, 2H), 7.23 (s, 1H), 7.11-7.09 (m, 5H), 6.98 (s, 1H), 4.12 (t, J = 2.4 Hz, J = 2.8 Hz, 2H), 1.89 (m, 2H), 1.63-1.35 (m, 10H), 0.95 (t, 3H). MALDI-TOF-MS, m/z: calcd for C27H28BBrF2N2OS [M]+: 556.117; found 556.073。
BDP1的合成
在100 mL 單口瓶中,依次加入中間體1 (96 mg, 0.3 mmol) 、 中間體5 (140 mg, 0.25 mmol)、四(三苯基膦)鈀 (11 mg, 0.01 mmol)、甲苯 (30 mL) 和碳酸鉀溶液 (20 mL 2M),排除空氣,氬氣保護,混合物在分別在70℃、90℃、110℃下反應12h、24h、36h後停止反應。將反應混合液冷卻至室溫,倒入100 mL 蒸餾水中,用乙酸乙酯萃取數次,有機層再用飽和食鹽水洗滌 (50 mL×3),合併有機相,無水硫酸鎂乾燥。減壓下旋轉蒸發去除溶劑。殘餘物以石油醚:乙酸乙酯 = 10: 1 (v/v)為洗脫劑用矽膠 (300-400目) 進行柱層析分離提純,得到墨綠色固體204 mg,產率61%。1H NMR (600 MHz, CDCl3), δ: 8.35 (s, 1H), 8.16 (s, 1H), 7.72 (d, J = 7.6 Hz, 2H), 7.64 (d, J = 8.6 Hz, 2H), 7.58 (s, 1H), 7.52 (d, J = 9.2 Hz, 1H), 7.44 (d, J = 6.7 Hz, 1H), 7.36–7.31 (m, 2H), 7.22 (d, J = 5.0 Hz, 1H), 7.20–7.16 (m, 2H), 7.12 (d, J = 8.6 Hz, 2H), 7.06–7.02 (m, 1H), 7.00 (s, 1H), 4.10 (t, J = 6.5 Hz, 2H), 1.90–1.85 (m, 2H), 1.51 (s, 7H), 1.43–1.26 (m, 11H), 0.91 (t, J = 6.9 Hz, 3H). 13C NMR (151 MHz, CDCl3), δ: 161.92, 154.47, 153.78, 146.41, 142.03, 140.53, 132.48, 127.91, 127.39, 127.08, 125.23, 124.17, 123.20, 122.63, 120.51, 120.03, 119.62, 114.78, 68.44, 46.93, 31.83, 29.46, 27.17, 26.08, 22.68, 14.13. MALDI-TOF-MS, m/z: calcd for C42H41BF2N2OS [M]+:670.300; found 670.209. HRMS, m/z: calcd for C42H41BF2N2OS [M]+: 670.3001; found 670.2997。
BDP2的合成
向100ml圓底燒瓶中加入中間體2和中間體5,反應條件和合成BDP1的條件相同,純化得到墨綠色固體,產率63%。1H NMR (600 MHz, CDCl3), δ: 8.42 (s, 1H), 8.17 (s, 1H), 8.07 (d, J = 7.9 Hz, 2H), 7.66 (d, J = 8.6 Hz, 2H), 7.52 (s, 1H), 7.46 (t, J= 7.6 Hz, 1H), 7.40 (d, J = 8.1 Hz, 2H), 7.25–7.21 (m, 3H), 7.18 (d, J = 4.3 Hz, 1H), 7.12 (d, J = 8.6 Hz, 2H), 7.06–7.03 (m, 1H), 7.01 (s, 1H), 4.31 (t, J = 7.3 Hz, 2H), 4.11 (t, J = 6.5 Hz, 2H), 1.88 (td, J = 12.7, 6.8 Hz, 4H), 1.44–1.23 (m, 20H), 0.91 (t, J = 6.9 Hz, 3H), 0.86 (t, J = 7.0 Hz, 3H). 13C NMR (151 MHz, CDCl3), δ: 161.92, 146.35, 142.39, 141.05, 140.88, 140.34, 136.13, 132.51, 130.15, 127.92, 125.81, 124.14, 123.18, 120.86, 120.33, 116.87, 114.78, 108.80, 31.83, 29.39, 29.04, 27.35, 26.10, 22.65, 14.11. MALDI-TOF-MS, m/z: calcd for C47H52BF2N3OS [M]+: 755.389; found 755.345. HRMS, m/z: calcd for C47H52BF2N3OS [M]+: 755.3892; found 755.3883。
BDP3的合成
向100ml圓底燒瓶中加入中間體3和中間體5,反應條件和合成BDP1的條件相同,純化得到墨綠色固體,產率59%。1H NMR (400 MHz, CDCl3), δ: 8.03 (s, 2H), 7.56 (d, J = 8.5 Hz, 2H), 7.27 (d, J = 8.1 Hz, 13H), 7.02 (dd, J = 18.6, 8.4 Hz, 12H), 6.93 (d, J = 8.6 Hz, 4H), 4.06 (t, J = 6.5 Hz, 2H), 1.71 (s, 8H), 1.55 (s, 11H), 1.28 (d, J = 19.8 Hz, 18H), 0.75 (s, 42H). MALDI- TOF-MS, m/z: calcd for C61H74BF2N3OS [M]+: 945.561; found 945.258. HRMS, m/z: calcd for C61H74BF2N3OS [M]+: 945.5614; found 945.5611。
上述實施例中目標產物BDP1-3在CH2Cl2溶液中和固體膜上的紫外可見吸收光譜和螢光光譜結果見表1,實施例中目標產物BDP1-3電化學性質的相關數據見表2。
。
a measured in CH2Cl2 solution. b measured in the neat film。
表1結果顯示,在CH2Cl2溶液中,幾種目標產物在620 nm左右均有較強的紫外吸收峰。BDP1和BDP2的最大吸收峰幾乎處於同一波長(620 nm),且吸收峰輪廓也相似,而BDP3的最大吸收峰則相對更趨向於長波方向(649 nm),吸收峰輪廓也更寬闊。這主要是因為三苯胺擁有更強的給電子能力,其與BODIPY核之間的D-A效應更為強烈,引起的ICT也更為明顯,降低了分子的能隙。BDP 1-3在溶液中均表現出良好的光譜吸收強度(6.8×104 -1.1×105M-1 cm-1),這可使分子高效利用光譜響應範圍內的光子轉換為光電流,具備良好的光伏電池材料特徵。
相對於溶液中的光譜吸收,BDP 1-3在固態下的光譜吸收均表現出5-9 nm的紅移,而且光譜吸收輪廓變得更為寬闊,吸收邊界擴展到750-850 nm的區間。這是由於有機分子在薄膜中比在溶液中有更好的共面性,共軛效應更強,形成更加緊密的π-π堆積所致。
表2結果顯示,幾種染料的第一氧化電位分別是1.19、1.10和0.96 eV,計算得BDP1-3對應的HOMO能級分別為:-5.45,-5.37和 -5.23 eV;LUMO能級分別為:-3.45,-3.37和 -3.32 eV。可以看到這幾個分子的HOMO能級均低於-5.2eV,因此幾個分子在空氣中具有較好的穩定性(-5.2eV是有機分子能夠穩定存在的能級,低於這個數值分子在空氣中能夠穩定存在),適合於做有機小分子太陽能電池給體材料。
實施例2
中間體1的合成:
在2-碘-9,9』-二甲基芴 (3.2 g, 10 mmol)、雙聯頻哪醇硼酯 (2.79 g, 11 mmol)、乙酸鉀 (2.94 g, 30 mmol)、DMF (80 mL)的混合液中加入Pd(dppf)Cl2 (360 mg, 0.5 mmol)。體系在氬氣保護下, 90oC反應8 h。停止反應,混合物冷卻至室溫後倒入200 mL 水中,用乙酸乙酯萃取 (50 mL × 3),再用飽和食鹽水洗滌數次,合併有機相,無水硫酸鎂乾燥。減壓旋轉蒸發,去除溶劑,殘餘物以石油醚:乙酸乙酯 = 20:1 (v/v) 為洗脫劑用矽膠 (200-300目) 進行柱層析分離,得到白色固體2.82 g,產率88%。1H NMR (600 MHz, CDCl3) δ: 7.91 (s, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.76 (t, J = 6.8 Hz, 2H), 7.48-7.45 (m, 1H), 7.37-7.34 (m, 2H), 1.53 (s, 6H), 1.40 (s, 12H) 。
中間體2的合成:
在3-溴-9-辛基咔唑 (2 g, 5.6 mmol)、雙聯頻哪醇硼酯 (1.7 g, 6.7 mmol)、乙酸鉀 (2.7 g, 28 mmol)、DMF (80 mL)的混合液中加入Pd(dppf)Cl2 (150 mg, 0.21 mmol)。體系在氬氣保護下, 90oC反應8 h。停止反應,混合物冷卻至室溫後倒入200 mL 水中,用二氯甲烷萃取 (50 mL × 3),再用飽和食鹽水洗滌數次,合併有機相,無水硫酸鎂乾燥。減壓旋轉蒸發,去除溶劑,殘餘物以石油醚:乙酸乙酯 = 20:1 (v/v) 為洗脫劑用矽膠 (200-300目) 進行柱層析分離,得到白色固體1.7 g,產率76%。
中間體3的合成:
中間體3 的反應條件與合成中間體2的條件類似,用4-溴-N,N-二(4-異辛基苯基)苯胺作為反應底物。純化後得到白色固體, 產率84%。
中間體4的合成:
在100 mL單口瓶中加入對辛氧基苯甲醛 (2.34 g, 10 mmol) 和新蒸的吡咯 (30 mL, 430 mmol), 向溶液中鼓氬氣置換10 min,在氬氣保護下迅速加入催化劑InCl3 (0.11 g, 0.5 mmol), 在室溫下磁力攪拌5 h, 向其中加入NaOH (0.2 g, 5 mmol)粉末繼續攪拌30 min,終止反應。減壓蒸餾,回收多餘的吡咯,得粗產品,用矽膠(200-300目)柱層析[洗脫液,V(石油醚):V(乙酸乙酯) = 7:1],純化得到白色晶體2.45 g, 產率70%。
在500 mL三口瓶中,取上述合成的白色晶體 (3.50 g, 10 mmol) 溶解於二氯甲烷 (80 mL) 中,加入四氯苯醌 (2.9 g, 12 mmol), 室溫條件下磁力攪拌,充分氧化8 h。然後將反應混合液置於氬氣保護下,慢慢滴加三氟化硼乙醚 (37 mL, 300 mmol),攪拌反應10 min 後再緩慢加入三乙胺 (41.7 mL, 300 mmol),滴加完畢後繼續反應8 h。混合物倒入氫氧化鈉溶液中,用二氯甲烷萃取。有機相用無水硫酸鈉乾燥。過濾、濾液減壓下去除溶劑。粗產品用矽膠 (200-300目) 柱層析[洗脫液,V(石油醚):V(乙酸乙酯)=10:1] 分離得紅綠色粉末狀化合物2.50 g,產率63%。
向100 mL三口瓶中依次加入上一步合成的紅綠色固體 (1.0 g, 2.5 mmol)、DMF (20 mL) 和二氯甲烷 (20 mL),在室溫下磁力攪拌幾分鐘後,裝上恆壓滴液漏鬥,抽真空,通入氬氣保護,將NBS (0.89 g, 5.0 mmol) 溶解在DMF (5 mL) 和二氯甲烷 (5 mL)中,通過恆壓滴液漏鬥逐滴滴入到三口瓶中,滴加完畢後再繼續反應4 h,整個過程避光。向三口瓶中加入50 mL去離子水,用二氯甲烷萃取三次 (80 mL × 3),有機層再經飽和食鹽水洗滌 (100 mL × 3),合併有機相,無水硫酸鎂乾燥過夜。過濾收集濾液,減壓下旋轉蒸發去除溶劑,得粗產品,用矽膠(200-300目)柱層析[洗脫液,V(石油醚):V(乙酸乙酯) = 20:1]純化得紅色粉末狀固體 0.97 g,產率70%。
中間體5的合成:
在100 mL單口瓶中,依次加入中間體4 (0.55 g, 1.0 mmol)、2-(三丁基錫)噻吩(0.45 g, 1.2 mmol), 氟化銫 (0.3 g, 2.0 mmol) 和甲苯 (30 mL),抽真空,再向反應瓶中加入四(三苯基膦)鈀 (0.15 g, 0.13 mmol),氬氣保護,120oC下磁力攪拌反應16 h。停止反應,冷卻至室溫,倒入水中,用乙酸乙酯萃取,再用飽和食鹽水洗滌 (30 mL × 3),收集有機相,無水硫酸鈉乾燥。減壓下旋轉蒸發去除溶劑,得粗產品,用矽膠 (300-400目) 柱層析 [洗脫液,V(石油醚):V(乙酸乙酯) = 10:1],得到褐色粉末狀化合物固體0.31 g,產率56%。
BDP1的合成
在100 mL 單口瓶中,依次加入中間體1 (96 mg, 0.3 mmol) 、 中間體5 (140 mg, 0.25 mmol)、四(三苯基膦)鈀 (11 mg, 0.01 mmol)、甲苯 (30 mL) 和碳酸鉀溶液 (20 mL 2M),排除空氣,氬氣保護,混合物在分別在65℃、85℃、115℃下反應12h、24h、34h後停止反應。將反應混合液冷卻至室溫,倒入100 mL 蒸餾水中,用乙酸乙酯萃取數次,有機層再用飽和食鹽水洗滌 (50 mL×3),合併有機相,無水硫酸鎂乾燥。減壓下旋轉蒸發去除溶劑。殘餘物以石油醚:乙酸乙酯 = 10: 1 (v/v)為洗脫劑用矽膠 (300-400目) 進行柱層析分離提純,得到墨綠色固體204 mg,產率61%。
BDP2的合成
向100ml圓底燒瓶中加入中間體2和中間體5,反應條件和合成BDP1的條件相同,純化得到墨綠色固體,產率63%。
BDP3的合成
向100ml圓底燒瓶中加入中間體3和中間體5,反應條件和合成BDP1的條件相同,純化得到墨綠色固體,產率59%。
本發明通過上述實施例來說明本發明的詳細合成方法,但本發明並不局限於上述方法,即不意味著本發明必須依賴上述反應條件才能實施。所屬技術領域的技術人員應該明了,對本發明的任何改進,對本發明反應溶劑催化劑的等效替換及反應具體條件的改變等,均落在本發明的保護範圍和公開範圍之內。