用於標記無載體[*I]MIBG的藥盒及其製備方法與流程
2023-05-02 13:30:26 1
本發明屬於放射性藥物及核醫學技術領域,具體涉及一種用於標記無載體[*I]MIBG的藥盒及其製備方法。
背景技術:
間碘苄胍(MIBG)屬於胍乙啶的類似物,可在腎上腺髓質中濃集,是降甲腎上腺素神經遞質(NE)的一種同系物,藉助於NE,可以被富含交感神經的細胞所吸收,是一種在診斷和治療心肌異常及神經內分泌瘤方面非常寶貴的載體。現有技術中廣泛使用以放射性碘如131I標記的MIBG進行顯像和神經內分泌瘤的診斷和治療。現在國內外臨床上用的[*I]MIBG多數是通過*I與MIBG用同位素交換的方法製得的,如加熱熔融法、(NH4)2SO4相交換碘化技術、Cu(Ⅱ)催化131I–MIBG的交換反應和亞銅催化法,並製備凍幹藥盒供臨床使用。由於該製劑以MIBG為標記前體,因其無放射性,運輸和使用方便,易於臨床推廣,避免了放射性物質在運輸、儲存中對環境的汙染風險,降低了放射性藥物的質量風險,減少同位素的衰變,降低了成本,因此使用MIBG為標記前體的藥盒通過同位素交換的方法製備[*I]MIBG有發展前途。但是,上述製劑的標記前體MIBG藥盒也存在諸多不足,由於用同位素交換技術製備的*I-MIBG標記率比較低,如131I-MIBG的一般標記率僅為50%-60%,因此製成的[*I]MIBG還需要進行複雜繁瑣的後處理步驟,通過陰離子樹脂柱層析或用無機分離劑AgO2來吸附游離的放射性碘,對製劑人員和設備條件等要求較高,因此一般醫院並不具備相應的製備條件,從而制約了*I-MIBG的臨床使用;另外,採用標記前體MIBG藥盒用同位素交換技術製備的*I-MIBG中,含有大量未標記的MIBG載體,然而大量臨床試驗證實,未標記的MIBG載體含量越低,[*I]MIBG會更有效且副作用更小,因此由於其標記率較低,該標記前體MIBG不適合於製作藥盒之用。為了提高[*I]MIBG的藥效和降低可能的副作用,對於不含有或含有較低量的未標記的MIBG載體的[*I]MIBG稱之為無載體[*I]MIBG,所述的無載體[*I]MIBG因標記率高,且藥效高而副作用小而被廣泛的研究及使用。Hunter課題組報導了在合成樹脂上鍵合錫基團的方法製備無載體[*I]MIBG(其合成路線見圖6所示),該方法製得的無載體[*I]MIBG簡單過濾即可分離,而且無多餘雜質,但是合成樹脂的方法和檢測比較複雜,不適用於常規實驗室的製備,因此其[*I]MIBG標記前體也不適合於製作藥盒之用。現有技術中還報導了以間位三甲基矽基苄胍為前體合成無載體[*I]MIBG的方法,但該方法合成步驟繁多,得到的無載體[*I]MIBG不穩定,而且在室溫條件下分解較快,使得藥物中依然含有大量的未標記的MIBG載體,使得製備得到的無載體[*I]MIBG藥效降低且副作用大增,同時溶液中游離的131I隨著時間延長而增加,而131I具有較大的毒性,使得[*I]MIBG不便於運輸、貯存和使用。又如專利文獻US2011040119A1中公開了一種以N,N-二叔丁氧羰基,3-三丁基錫基苄胍為前體合成無載體[*I]MIBG的方法,該方法在3-三丁基錫基苄胍前體中引入了BOC保護基團,雖然標記率較高,但是已標記的[*I]MIBG中帶入了雜質,不利於[*I]MIBG的使用。因此上述的[*I]MIBG標記前體同樣不適合於製作藥盒之用。
技術實現要素:
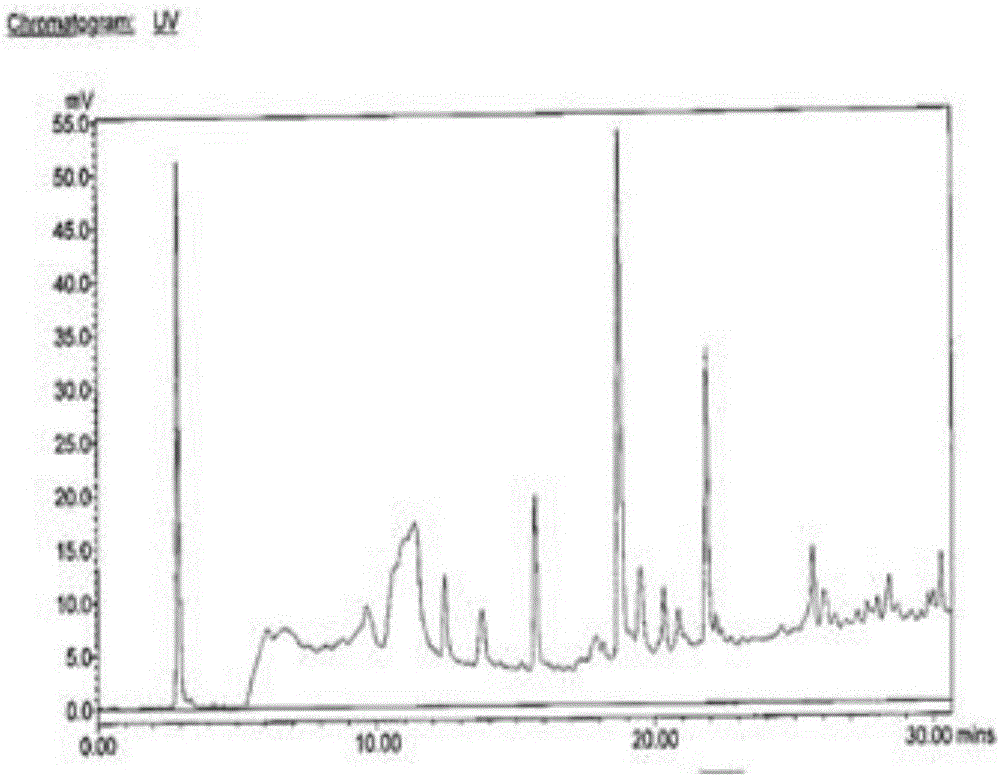
為此,本發明所要解決的技術問題在於現有技術中製備的[*I]MIBG標記率低、雜質多和不穩定,進而提供一種用於製備具有標記率高、雜質少和穩定的[*I]MIBG的藥盒及其製備方法。為解決上述技術問題,本發明所述的用於標記無載體[*I]MIBG的藥盒,每套所述藥盒包括各自獨立包裝的KitA、KitB和KitC部分,其中,所述KitA部分包括:MSnBG原料藥凍乾粉末1-50μg以及調節所述MSnBG原料藥水溶液pH為3.5-8.0的pH緩衝劑凍乾粉末適量;所述KitB部分包括:氧化劑凍乾粉末10-1000μg;所述KitC部分包括:還原劑凍乾粉末6-600μg。上述MSnBG原料藥為1-[4-(三烷基甲錫烷基)苄基]胍,其為1-[4-(三甲基甲錫烷基)苄基]胍、1-[4-(三乙基甲錫烷基)苄基]胍、1-[4-(三丙基甲錫烷基)苄基]胍或1-[4-(三丁基甲錫烷基)苄基]胍中的一種。在所述的用於標記無載體[*I]MIBG的藥盒中,每套所述藥盒包括,所述KitA的部分包括:MSnBG凍幹25μg以及調節所述MSnBG原料藥水溶液pH為6.5的pH緩衝劑凍乾粉末適量;所述KitB的部分包括:氧化劑凍乾粉末300μg;所述KitC的部分包括:還原劑凍乾粉末200μg。在所述的用於標記無載體[*I]MIBG的藥盒中,所述氧化劑為氯胺T、二氯胺T、氯胺B或1,3,4,6-四氯-3α,6α-二苯基甘脲中的一種。在所述的用於標記無載體[*I]MIBG的藥盒中,所述還原劑為焦亞硫酸鈉。在所述的用於標記無載體[*I]MIBG的藥盒中,所述pH緩衝劑為磷酸氫二鉀和氫氧化鈉的混合物、醋酸銨和鹽酸混合物、磷酸二氫鈉和磷酸氫二鈉的混合物或者磷酸氫二鉀和磷酸二氫鉀的混合物中的一種。本發明提供了一種配製所述的用於標記無載體[*I]MIBG的藥盒的方法,包括如下步驟:A,稱取選定重量的MSnBG原料藥溶於1-2mL蒸餾水中,加入適量所述pH緩衝劑調至選定的pH值,冷凍乾燥至其內部物質呈白色粉末狀,得到所述的KitA部分;其中所述蒸餾水優選為1mL;B,稱取選定重量的氧化劑溶於1-2mL蒸餾水中,冷凍乾燥至其內部物質呈白色粉末狀,得到所述的KitB部分;其中所述蒸餾水優選為1mL;C,稱取選定重量的還原劑溶於1-2mL蒸餾水中,冷凍乾燥至其內部物質呈白色粉末狀,得到所述的KitC部分;其中所述蒸餾水優選為1mL。在所述的用於標記無載體[*I]MIBG的藥盒的配製方法中,所述冷凍乾燥步驟具體為:控制溫度為-35~-25℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-25~-15℃,冷凍乾燥3小時;再將所述溫度調節至-15~-5℃,冷凍乾燥12小時;再將所述溫度調節至-5~5℃,冷凍乾燥1小時;再將溫度調節至5~15℃,乾燥2小時;再將所述溫度調節至20~30℃,乾燥1.5小時;控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得凍乾粉末。優選的,在所述的用於標記無載體[*I]MIBG的藥盒的配製方法中,所述冷凍乾燥步驟具體為:控制溫度為-30℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-20℃,冷凍乾燥3小時;再將所述溫度調節至-10℃,冷凍乾燥12小時;再將所述溫度調節至0℃,冷凍乾燥1小時;再將溫度調節至10℃,乾燥2小時;再將所述溫度調節至25℃,乾燥1.5小時本發明提供了一種利用所述藥盒標記無載體[*I]MIBG的方法,向所述kitB部分中的氧化劑凍乾粉末中加入0.1-5.0mL的0.1-50mCi的含放射性同位素*I離子的水溶液中溶解混勻,隨後將上述溶解液加入至所述kitA部分中混勻,常溫條件下反應8-12min,隨後將所述kitC部分中的凍乾粉末加入至上述反應液中混勻,常溫條件下反應8-12min,得到標記的無載體[*I]MIBG。優選的,加入的所述放射性同位素*I離子水溶液為1mL的5mCi的*I離子的水溶液。所述放射性同位素*I離子的水溶液可以為123I、124I、125I或131I離子的水溶液。本發明提供的所述的用於標記無載體[*I]MIBG的藥盒在腫瘤顯像的領域的應用。本發明提供的利用所述藥盒標記無載體[*I]MIBG的方法在腫瘤顯像的領域的應用本發明的上述技術方案相比現有技術具有以下優點:(1)本發明所述的用於標記無載體[*I]MIBG的藥盒,每套藥盒中包括各自獨立包裝的KitA、KitB和KitC部分,KitA的部分包括:MSnBG原料藥凍乾粉末1-50μg以及調節所述MSnBG原料藥水溶液pH為3.5-8.0的pH緩衝劑凍乾粉末適量;KitB的部分包括:氧化劑凍乾粉末10-1000μg;KitC的部分包括:還原劑凍乾粉末6-600μg,利用上述藥盒製備得到的[*I]MIBG具有標記率高、雜質少和穩定等優點,標記率平均達到了90%以上,放射化學純度平均達到了98%,利於臨床上的推廣使用,解決了現有技術中製備的[*I]MIBG標記率低、雜質多和不穩定的問題;(2)本發明所述的利用所述藥盒標記無載體[*I]MIBG的方法,製備的[*I]MIBG標記率平均高達90%以上,且方法步驟簡單易操作,利於臨床的應用。附圖說明為了使本發明的內容更容易被清楚的理解,下面根據本發明的具體實施例並結合附圖,對本發明作進一步詳細的說明,其中圖1是本發明實施例6的空白標記樣品的紫外HPLC的圖譜;圖2是本發明實施例6的無載體[131I]MIBG樣品溶液的紫外HPLC的圖譜;圖3是本發明實施例6的空白標記樣品的放射性HPLC的圖譜;圖4是本發明實施例6的無載體[131I]MIBG樣品溶液的放射性HPLC的圖譜;圖5是本發明實施例中所述的MSnBG的合成路線圖;圖6是在合成樹脂上鍵合錫基團的方法製備無載體[*I]MIBG的合成路線圖。具體實施方式本發明的下述實施例中的所述MSnBG原料藥可選用市售產品進行凍乾粉末製備,也可以按照現有文獻記載進行常規製備,例如採用如下方程式所述方法製備得到:(1)所述式A-I所示的間碘苄胍的碳酸氫鹽可以為市售常規產品為原料,也可以以間碘苄胺鹽酸鹽為原料進行常規的製備,具體的製備步驟可以參照專利文獻US2011040119A1中公開的所述式A-I所示的間碘苄胍的碳酸氫鹽的合成方法製備得到,所得產品進行核磁和MR檢測,其1HNMR(500M,CD3OD)δ4.34(s,2H),7.10-7.14(t,1H),7.34-7.36(d,1H),7.63-7.65(d,1H),7.69(s,1H),與所述間碘苄胍的碳酸氫鹽標準圖譜吻合;(2)所述式A-II所示的1-[4-(三烷基甲錫烷基)苄基]胍的合成,取500mg(1.48mmol)的所述式A-I所示的間碘苄胍的碳酸氫鹽溶於10mL的DMSO的溶液中,隨後加入947mg(1.63mmol)的雙(三丁基錫)混勻,並整體溶解於30mL的1,4-二氧六環中,隨後加入104mg(0.15mmol)的雙三苯基磷二氯化鈀,控制溫度100℃下恆溫進行stille有機錫反應至溶液變黑,至溶液顏色不再加深,旋幹製得的上述溶液,取乙酸乙酯溶解旋幹後的殘留物,隨後水洗所述溶解液,再次旋幹,取甲醇溶解再次旋幹後的殘留物,隨後用正己烷洗所述溶解液,然後乾燥,所述式A-II所示的1-[4-(三烷基甲錫烷基)苄基]胍採用製備型色譜進行分離,所述色譜條件為:色譜柱:C18反相色譜柱,粒徑5um,柱子規格4.6*250mm;洗脫:以甲醇-水為流動相進行梯度洗脫,具體洗脫程序為0-15min時,甲醇-水的體積比由50%:50%→100%:0;運行溫度為25℃;流速為0.01L/min;檢測波長為230nm;收集出峰時間為13-15min下的流出液,收集含有式A-II所示的1-[4-(三烷基甲錫烷基)苄基]胍的流份;分離得淡黃色液體,所得產品進行核磁和MR檢測,顯示1HNMR(500M,CD3OD)δ0.88-0.91(m,9H),1.08-1.11(m,6H),1.32-1.37(m,6H),1.55-1.58(m,6H),4.39(s,2H),7.24-7.26(m,1H),7.33-7.37(m,1H),7.41(m,2H);MR[MH]+:439,與1-[4-(三烷基甲錫烷基)苄基]胍標準圖譜吻合,即所述式A-II所示化合物為1-[4-(三烷基甲錫烷基)苄基]胍。進一步檢測其純度95%,產率為48.3%。上述雙(三丁基錫)可以替換成雙(三甲基錫)、雙(三乙基錫)和雙(三丙基錫)分別製備1-[4-(三甲基甲錫烷基)苄基]胍、1-[4-(三乙基甲錫烷基)苄基]胍和1-[4-(三丙基甲錫烷基)苄基]胍。本發明下述實施例中的MSnBG的合成路線如圖5所示。下述各實施例中所涉及使用的儀器型號包括:HPLC,型號為waters泵1525,產自美國;C18反相柱,規格為hypersilODS2,5μm,4.6×150mm,產自依利特公司,中國;放射性活度計,型號為CALIRADISOTOPECALIBRATORCRC-250,產自VICTOREEN公司,美國;γ-計數器,型號為γ-counter2480,產自PE公司,美國;紫外檢測器2487產自waters公司;紅外光譜儀,型號為IRBrukerTENSOR-27型,產自德國;核磁共振儀,型號為BrukerAvanceⅢ400,產自德國;質譜儀,型號為SQDetector2,產自美國;同位素檢測器,型號為Radiomatic610TR,產自PE公司。實施例1本實施例所述的用於標記無載體[*I]MIBG的藥盒包括三個各自獨立包裝的KitA、KitB和KitC部分,其中,所述KitA的部分包括:MSnBG原料藥凍乾粉末1μg以及調節所述MSnBG原料藥水溶液pH為3.5的pH緩衝劑凍乾粉末適量;所述KitB的部分包括:二氯胺T凍乾粉末10μg;所述KitC的部分包括:焦亞硫酸鈉凍乾粉末6μg。上述藥盒按照以下配製方法製備得到,具體步驟如下:A,稱取所述MSnBG原料藥1μg溶於1mL蒸餾水中,分別加入醋酸銨為10mg、7M的鹽酸溶液0.0152mL,將上述MSnBG原料藥水溶液調至pH為3.5,然後將上述MSnBG原料藥水溶液按照以下步驟冷凍乾燥:控制溫度為-35℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-25℃,冷凍乾燥3小時;再將所述溫度調節至-15℃,冷凍乾燥12小時;再將所述溫度調節至-5℃,冷凍乾燥1小時;再將溫度調節至5℃,乾燥2小時;再將所述溫度調節至20℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitA部分;B,稱取所述二氯胺T10μg溶於1mL蒸餾水中,然後將上述溶液按照以下步驟冷凍乾燥:控制溫度為-35℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-25℃,冷凍乾燥3小時;再將所述溫度調節至-15℃,冷凍乾燥12小時;再將所述溫度調節至-5℃,冷凍乾燥1小時;再將溫度調節至5℃,乾燥2小時;再將所述溫度調節至20℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitB部分;C,稱取所述焦亞硫酸鈉6μg溶於1mL蒸餾水中,然後將上述溶液按照以下步驟冷凍乾燥:控制溫度為-35℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-25℃,冷凍乾燥3小時;再將所述溫度調節至-15℃,冷凍乾燥12小時;再將所述溫度調節至-5℃,冷凍乾燥1小時;再將溫度調節至5℃,乾燥2小時;再將所述溫度調節至20℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitC部分。實施例2本實施例所述的用於標記無載體[*I]MIBG的藥盒包括三個各自獨立包裝的KitA、KitB和KitC部分,其中,所述KitA的部分包括:MSnBG原料藥凍乾粉末25μg以及調節所述MSnBG原料藥水溶液pH為6.5的pH緩衝劑凍乾粉末適量;所述KitB的部分包括:氯胺T凍乾粉末300μg;所述KitC的部分包括:焦亞硫酸鈉凍乾粉末200μg。上述藥盒按照以下配製方法製備得到,具體步驟如下:A,稱取所述MSnBG原料藥25μg溶於1mL蒸餾水中,分別加入磷酸二氫鉀3.4mg、氫氧化鈉0.3mg,將上述MSnBG原料藥水溶液調至pH為6.5,然後將上述MSnBG原料藥水溶液按照以下步驟冷凍乾燥:控制溫度為-30℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-20℃,冷凍乾燥3小時;再將所述溫度調節至-10℃,冷凍乾燥12小時;再將所述溫度調節至0℃,冷凍乾燥1小時;再將溫度調節至10℃,乾燥2小時;再將所述溫度調節至25℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitA部分;B,稱取所述氯胺T300μg溶於1mL蒸餾水中,然後將上述溶液按照以下步驟冷凍乾燥:控制溫度為-30℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-20℃,冷凍乾燥3小時;再將所述溫度調節至-10℃,冷凍乾燥12小時;再將所述溫度調節至0℃,冷凍乾燥1小時;再將溫度調節至10℃,乾燥2小時;再將所述溫度調節至25℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitB部分;C,稱取所述焦亞硫酸鈉200μg溶於1mL蒸餾水中,然後將上述溶液按照以下步驟冷凍乾燥:控制溫度為-30℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-20℃,冷凍乾燥3小時;再將所述溫度調節至-10℃,冷凍乾燥12小時;再將所述溫度調節至0℃,冷凍乾燥1小時;再將溫度調節至10℃,乾燥2小時;再將所述溫度調節至25℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitC部分。實施例3本實施例所述的用於標記無載體[*I]MIBG的藥盒包括三個各自獨立包裝的KitA、KitB和KitC部分,其中,所述KitA的部分包括:MSnBG原料藥凍乾粉末50μg以及調節所述MSnBG原料藥水溶液pH為8.0的pH緩衝劑凍乾粉末適量;所述KitB的部分包括:1,3,4,6-四氯-3α,6α-二苯基甘脲凍乾粉末1000μg;所述KitC的部分包括:焦亞硫酸鈉凍乾粉末600μg。上述藥盒按照以下配製方法製備得到,具體步驟如下:A,稱取所述MSnBG原料藥50μg溶於2mL蒸餾水中,分別加入磷酸二氫鈉0.41mg、磷酸氫二鈉5.59mg,將上述MSnBG原料藥水溶液調至pH為8.0,然後將上述MSnBG原料藥水溶液按照以下步驟冷凍乾燥:控制溫度為-25℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-15℃,冷凍乾燥3小時;再將所述溫度調節至-5℃,冷凍乾燥12小時;再將所述溫度調節至5℃,冷凍乾燥1小時;再將溫度調節至15℃,乾燥2小時;再將所述溫度調節至30℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitA部分;B,稱取所述1,3,4,6-四氯-3α,6α-二苯基甘脲1000μg溶於2mL蒸餾水中,然後將上述溶液按照以下步驟冷凍乾燥:控制溫度為-25℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-15℃,冷凍乾燥3小時;再將所述溫度調節至-5℃,冷凍乾燥12小時;再將所述溫度調節至5℃,冷凍乾燥1小時;再將溫度調節至15℃,乾燥2小時;再將所述溫度調節至30℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitB部分;C,稱取所述焦亞硫酸鈉600μg溶於2mL蒸餾水中,然後將上述溶液按照以下步驟冷凍乾燥:控制溫度為-25℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-15℃,冷凍乾燥3小時;再將所述溫度調節至-5℃,冷凍乾燥12小時;再將所述溫度調節至5℃,冷凍乾燥1小時;再將溫度調節至15℃,乾燥2小時;再將所述溫度調節至30℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitC部分。實施例4本實施例所述的用於標記無載體[*I]MIBG的藥盒包括三個各自獨立包裝的KitA、KitB和KitC部分,其中,所述KitA的部分包括:MSnBG原料藥凍乾粉末50μg以及調節所述MSnBG原料藥水溶液pH為8.0的pH緩衝劑凍乾粉末適量;所述KitB的部分包括:氯胺B凍乾粉末500μg;所述KitC的部分包括:焦亞硫酸鈉凍乾粉末400μg。上述藥盒按照以下配製方法製備得到,具體步驟如下:A,稱取所述MSnBG原料藥50μg溶於1.5mL蒸餾水中,分別加入磷酸二氫鈉3.4mg、磷酸氫二鈉0.3mg,將上述MSnBG原料藥水溶液調至pH為6.5,然後將上述MSnBG原料藥水溶液按照以下步驟冷凍乾燥:控制溫度為-35℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-15℃,冷凍乾燥3小時;再將所述溫度調節至-15℃,冷凍乾燥12小時;再將所述溫度調節至5℃,冷凍乾燥1小時;再將溫度調節至5℃,乾燥2小時;再將所述溫度調節至20℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitA部分;B,稱取所述氯胺B500μg溶於1.5mL蒸餾水中,然後將上述溶液按照以下步驟冷凍乾燥:控制溫度為-25℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-23℃,冷凍乾燥3小時;再將所述溫度調節至-7℃,冷凍乾燥12小時;再將所述溫度調節至0℃,冷凍乾燥1小時;再將溫度調節至13℃,乾燥2小時;再將所述溫度調節至28℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitB部分;C,稱取所述焦亞硫酸鈉400μg溶於1.5mL蒸餾水中,然後將上述溶液按照以下步驟冷凍乾燥:控制溫度為-27℃,預凍3小時,隨後冷凍乾燥2.5小時;將所述溫度調節至-18℃,冷凍乾燥3小時;再將所述溫度調節至-13℃,冷凍乾燥12小時;再將所述溫度調節至3℃,冷凍乾燥1小時;再將溫度調節至8℃,乾燥2小時;再將所述溫度調節至23℃,乾燥1.5小時;其中,控制上述冷凍乾燥過程中真空低於5Pa時自動摻氣,當所述冷凍乾燥過程結束,關閉自動摻氣,並繼續乾燥2.5小時,隨後真空壓塞,軋蓋,即得內部物質呈白色粉末狀的凍乾粉末,得到所述的KitC部分。實施例5本實施例利用所述藥盒標記無載體[123I]MIBG的方法,具體步驟如下:取實施例1中所述的藥盒,向所述kitB部分中的氧化劑凍乾粉末中加入0.1mL的50mCi的含放射性同位素123I離子的水溶液中溶解混勻,隨後將上述溶解液加入至所述kitA部分中混勻,常溫條件下反應8min,隨後將所述kitC部分中的凍乾粉末加入至上述反應液中混勻,常溫條件下反應8min,得到標記的無載體[123I]MIBG。分別採用紫外HPLC法和放射性HPLC法檢測空白標記樣品和上述製備的無載體[123I]MIBG的樣品溶液,所述空白標記樣品溶液0.1-5mL的含放射性濃度範圍為0.1-141mCi/mL的Na123I、二氯胺T10μg、焦亞硫酸鈉6μg和緩衝劑pH為3.5的混合溶液,所述無載體[123I]MIBG的樣品溶液0.1-5mL的本實施例製備的無載體[123I]MIBG;所述HPLC的色譜條件為C18反相色譜柱,填料粒徑5μm,規格為4.6×150mm;以甲醇-水為流動相進行梯度洗脫,具體洗脫程序為0-30min時,甲醇-水的體積比為0:100%→100%:0,30-60min時甲醇-水的體積比為100%:0→100%:0,運行溫度為10℃~30℃;流速為1mL/min;檢測波長為254nm;收集出峰時間為19min下的流出液,所述空白標記樣品和上述製備的無載體[123I]MIBG的樣品溶液的檢測條件相同。本實施例利用所述藥盒標記得到的無載體[123I]MIBG的檢測結果如下:所述無載體[123I]MIBG的標記率為86.4%,純度為96.0%。實施例6本實施例利用所述藥盒標記無載體[131I]MIBG的方法,具體步驟如下:分別取實施例2中所述的藥盒,向所述kitB部分中的氧化劑凍乾粉末中加入1mL的5mCi的含放射性同位素131I離子的水溶液中溶解混勻,隨後將上述溶解液加入至所述kitA部分中混勻,常溫條件下反應10min,隨後將所述kitC部分中的凍乾粉末加入至上述反應液中混勻,常溫條件下反應10min,得到標記的無載體[131I]MIBG。分別採用紫外HPLC法和放射性HPLC法檢測空白標記樣品和上述製備的無載體[131I]MIBG的樣品溶液,所述空白標記樣品溶液0.1-5mL的含放射性濃度範圍為0.1-141mCi/mL的Na131I、氯胺T300μg、焦亞硫酸鈉200μg和緩衝劑pH為6.5的混合溶液,所述無載體[131I]MIBG的樣品溶液0.1-5mL的本實施例製備的無載體[131I]MIBG;所述HPLC的色譜條件為C18反相色譜柱,填料粒徑5μm,規格為4.6×150mm;以甲醇-水為流動相進行梯度洗脫,具體洗脫程序為0-30min時,甲醇-水的體積比為0:100%→100%:0,30-60min時甲醇-水的體積比為100%:0→100%:0,運行溫度為10℃~30℃;流速為1mL/min;檢測波長為254nm;收集出峰時間為19min下的流出液,所述空白標記樣品和上述製備的無載體[131I]MIBG的樣品溶液的檢測條件相同。本實施例利用實施例2中的所述藥盒標記得到的無載體[131I]MIBG的檢測結果如下:所述空白標記樣品和上述利用實施例2中所述藥盒標記得到的無載體[131I]MIBG的樣品溶液的紫外HPLC圖譜見圖1和圖2,所述空白標記樣品和上述利用實施例2中所述的藥盒標記得到的無載體[131I]MIBG的樣品溶液的放射性HPLC圖譜見圖3和圖4,圖3中空白標記樣品溶液中以131I-離子形式存在的,在RT=5min處有明顯的吸收峰,而在圖4中的無載體[131I]MIBG的樣品溶液的放射性HPLC圖譜中在RT=5min處沒有明顯的吸收峰,說明本實施利用實施例2中的所述藥盒標記無載體[131I]MIBG的方法中的131I-基本全部參加反應,製備的無載體[131I]MIBG中游離的131I-較少,在RT=19min處出現明顯的吸收峰,該吸收峰對應於其紫外HPLC圖譜上的同一保留時間處的吸收峰見圖2,因此可以得出,本實施例製備的[131I]MIBG標記率達到了90%以上,通過計算得其標記率為94.3%,放射化學純度為99.2%。實施例7本實施例利用所述藥盒標記無載體[125I]MIBG的方法,具體步驟如下:取實施例3中所述的藥盒,向所述kitB部分中的氧化劑凍乾粉末中加入5mL的0.1mCi的含放射性同位素125I離子的水溶液中溶解混勻,隨後將上述溶解液加入至所述kitA部分中混勻,常溫條件下反應12min,隨後將所述kitC部分中的凍乾粉末加入至上述反應液中混勻,常溫條件下反應12min,得到標記的無載體[125I]MIBG。分別採用紫外HPLC法和放射性HPLC法檢測空白標記樣品和上述製備的無載體[125I]MIBG的樣品溶液,所述空白標記樣品溶液0.1-5mL的含放射性濃度範圍為0-141mCi/mL的Na125I、1,3,4,6-四氯-3α,6α-二苯基甘脲1000μg、焦亞硫酸鈉600μg和緩衝劑pH為8.0的混合溶液,所述無載體[125I]MIBG的樣品溶液為0.1-5mL的本實施例製備的無載體[125I]MIBG;所述HPLC的色譜條件為C18反相色譜柱,填料粒徑5μm,規格為4.6×150mm;以甲醇-水為流動相進行梯度洗脫,具體洗脫程序為0-30min時,甲醇-水的體積比為0:100%→100%:0,30-60min時甲醇-水的體積比為100%:0→100%:0,運行溫度為10℃~30℃;流速為1mL/min;檢測波長為254nm;收集出峰時間為19min下的流出液,所述空白標記樣品和上述製備的無載體[125I]MIBG的樣品溶液的檢測條件相同。本實施例利用所述藥盒標記得到的無載體[125I]MIBG的檢測結果如下:所述無載體[125I]MIBG的標記率為94.2%,放射化學純度為99.1%。實施例8本實施例利用所述藥盒標記無載體[124I]MIBG的方法,具體步驟如下:取實施例4中所述的藥盒,向所述kitB部分中的氧化劑凍乾粉末中加入3mL的25mCi的含放射性同位素124I離子的水溶液中溶解混勻,隨後將上述溶解液加入至所述kitA部分中混勻,常溫條件下反應12min,隨後將所述kitC部分中的凍乾粉末加入至上述反應液中混勻,常溫條件下反應12min,得到標記的無載體[124I]MIBG。分別採用紫外HPLC法和放射性HPLC法檢測空白標記樣品和上述製備的無載體[124I]MIBG的樣品溶液,所述空白標記樣品溶液0.1-5mL的含放射性濃度範圍為0.1-141mCi/mL的Na124I、氯胺B500μg、焦亞硫酸鈉400μg和緩衝劑pH為6.5的混合溶液,所述無載體[124I]MIBG的樣品溶液為0.1-5mL的本實施例製備的無載體[124I]MIBG;所述HPLC的色譜條件為C18反相色譜柱,填料粒徑5μm,規格為4.6×150mm;以甲醇-水為流動相進行梯度洗脫,具體洗脫程序為0-30min時,甲醇-水的體積比為0:100%→100%:0,30-60min時甲醇-水的體積比為100%:0→100%:0,運行溫度為10℃~30℃;流速為1mL/min;檢測波長為254nm;收集出峰時間為19min下的流出液,所述空白標記樣品和上述製備的無載體[124I]MIBG的樣品溶液的檢測條件相同。本實施例利用所述藥盒標記得到的無載體[124I]MIBG的檢測結果如下:所述無載體[124I]MIBG的標記率為90.7%,放射化學純度為98.0%。效果對照例按照現有技術中的專利文獻US2011040119A1中公開的合成方法製備得到[*I]MIBG。計算所述[*I]MIBG的標記率為80%。綜上可見,本發明所述的用於標記無載體[*I]MIBG的藥盒標記無載體[*I]MIBG的標記率明顯高於現有技術中專利文獻US2011040119A1中得到的[*I]MIBG的標記率,並且上述專利文獻US2011040119A1中得到的[*I]MIBG引入了更多的雜質,不利於提純和後續藥用研究。顯然,上述實施例僅僅是為清楚地說明所作的舉例,而並非對實施方式的限定。對於所屬領域的普通技術人員來說,在上述說明的基礎上還可以做出其它不同形式的變化或變動。這裡無需也無法對所有的實施方式予以窮舉。而由此所引伸出的顯而易見的變化或變動仍處於本發明創造的保護範圍之中。