一種用於檢測鋅離子的螢光探針及其製備方法與應用與流程
2023-07-31 05:00:01 4
本發明屬於有機合成與分析化學技術領域,具體涉及一種用於檢測鋅離子的螢光探針及其製備方法與應用。
背景技術:
由於螢光探針的高選擇性和靈敏度,現已廣泛應用於陽離子和陰離子的檢測當中。這些螢光探針很容易被修飾,以適應不同的測試環境和測試物。目前,二甲基吡咯bodipy已成為了一個很有吸引力的螢光探針之一。這種bodipy螢光探針一般光化學穩定性強,螢光強度高,環境適應力強,展現出很好的應用前景。
鋅離子在人體內酶的活性中起著重要作用,它不僅參與上皮組織分化,還參與金屬硫蛋白的轉運和儲存;不僅有抗炎作用,還能夠降低紫外誘導細胞和基因損傷;不僅能提高皮膚纖維細胞對氧化的耐受力,還能提高皮膚彈性和減少皺紋。但是,鋅攝入量過多,會導致中毒,引發腹痛、嘔吐等急性鋅中毒症狀,還會導致腎、肝功能免疫力受損。所以,在環境當中能快速簡便地檢測鋅離子顯得尤為重要。
技術實現要素:
本發明的一個目的在於提供一種能夠用於鋅離子檢測的螢光探針,通過明顯的螢光猝滅可以靈敏地檢測到鋅離子的存在,有很好的應用前景。
本發明所述的一種用於檢測鋅離子的螢光探針,其化學名為8-[4-(1-氧代羧戊氧基-乙氧基)苯基]-4,4-二氟-1,3,5,7-四甲基-4-硼-3a,4a-二吡咯(簡稱羧基bodipy),該螢光探針是一種鏈端帶羧基的多烷氧基二甲基吡咯bodipy的結構,其具體結構為:
本發明另一目的在於提供所述螢光探針(羧基bodipy)的製備方法。
本發明所述羧基bodipy合成路線如下:
該羧基bodipy的製備方法,具體包括以下步驟:
1)將對羥基苯甲醛與氯乙醇以摩爾比1:1~1:10比例,在碳酸鉀與乙腈體系中加熱攪拌回流,反應10~20小時,反應產物經處理後得到化合物1,所述化合物1為4-(2-羥乙氧基)苯甲醛,化合物1的結構式如下:
2)將化合物1與戊二酸酐以摩爾比1:1~1:20比例,在甲苯中加熱回流20~40小時,反應產物經處理後得到化合物2,所述化合物2的為4-(1-氧代羧戊氧基-乙氧基)苯甲醛,化合物2的結構式如下:
3)在氮氣保護下,將2,4-二甲基吡咯與化合物2以摩爾比1:1~5:1比例混合加入ch2cl2,常溫反應3~12小時;
接著加入2,3-二氯-5,6-二氰對苯醌(簡稱ddq),其中ddq與化合物2的摩爾比為4:1~1:4,常溫反應2~12小時;
然後向混合體系中依次加入三乙胺和三氟化硼乙醚,其中,三乙胺與化合物2的摩爾比為50:1~10:1,三氟化硼乙醚與化合物2的摩爾比為50:1~10:1,室溫反應2~12個小時;
反應結束後,產物用蒸餾水洗滌,用二氯甲烷萃取,硫酸鎂乾燥,過濾,濃縮濾液,經矽膠柱層析分離產物,減壓旋幹後得到棗紅色固體,即為所述螢光探針。
本發明的一個較優公開例中,所述螢光探針合成反應步驟3)中,2,4-二甲基吡咯與化合物2的摩爾比為2:1,常溫反應8小時。
本發明的一個較優公開例中,所述螢光探針合成反應步驟3)中,ddq與化合物2的摩爾比為1:1,常溫反應6小時。
本發明的一個較優公開例中,所述螢光探針合成反應步驟3)中,三乙胺與化合物2的摩爾比為22:1,三氟化硼乙醚與化合物2的摩爾比為20:1,室溫反應5-6個小時。
本發明製得的螢光探針羧基bodipy,其分子式為c26h29bf2n2o5,紅外光譜(kbr),v/cm-1:2923,2847(c-h),1729,1706(o=c),1305(c-n),1245(ar-o-c).核磁氫譜(400mhz,cdcl3)δ7.36(s,1h,oh),7.18(d,j=8.0hz,2h),7.03(d,j=8.0hz,2h),5.99(s,2h),4.50(t,j=6.4hz,2h),4.23(t,j=6.4hz,2h),2.56(s,3h),2.40-2.50(m,4h),1.98-2.02(m,2h),1.42(s,3h).高分辨質譜(m/s):計算值c26h29bf2n2o5498.2139(m)+,測量值498.2144。
本發明所製備的螢光探針羧基bodipy在溶液中顯淺黃色,在507nm處有較強螢光發射,螢光量子產率0.96,水溶性好,毒性低,該螢光探針在溶液中與鋅離子形成1:1絡合物,並導致螢光發生明顯猝滅,可用於環境中鋅離子的靈敏檢測,其它離子幹擾小,是一種理想的鋅離子快速檢測傳感器。
本發明所製備的羧基bodipy可對鋅離子進行定性、定量的檢測,將濃度呈梯度變化的鋅離子溶液與羧基bodipy溶液混合後,測定相應的螢光強度,然後以鋅離子的濃度為橫坐標,混合體系的螢光強度為縱坐標作圖,可根據螢光強度從圖中讀出待測溶液中的鋅離子濃度。
本發明的有益成果:所製備的羧基bodipy在鋅離子存在下螢光發生顯著改變,其它離子的存在不幹擾鋅離子的檢測,檢測限為3.64μm,可用於高選擇性高靈敏性地檢測鋅離子,這對於複雜環境中鋅離子的檢測有重要的現實應用價值。
附圖說明
圖1為羧基bodipy在二甲亞碸比水為9:1的溶液中,10-6mol/l羧基bodipy與10-4mol/l各離子的紫外吸收圖譜,橫坐標為波長、縱坐標為吸光度。紫外光譜吸光度對鋅離子的變化最為明顯,表明羧基bodipy對鋅離子有較強的螢光識別響應能力。
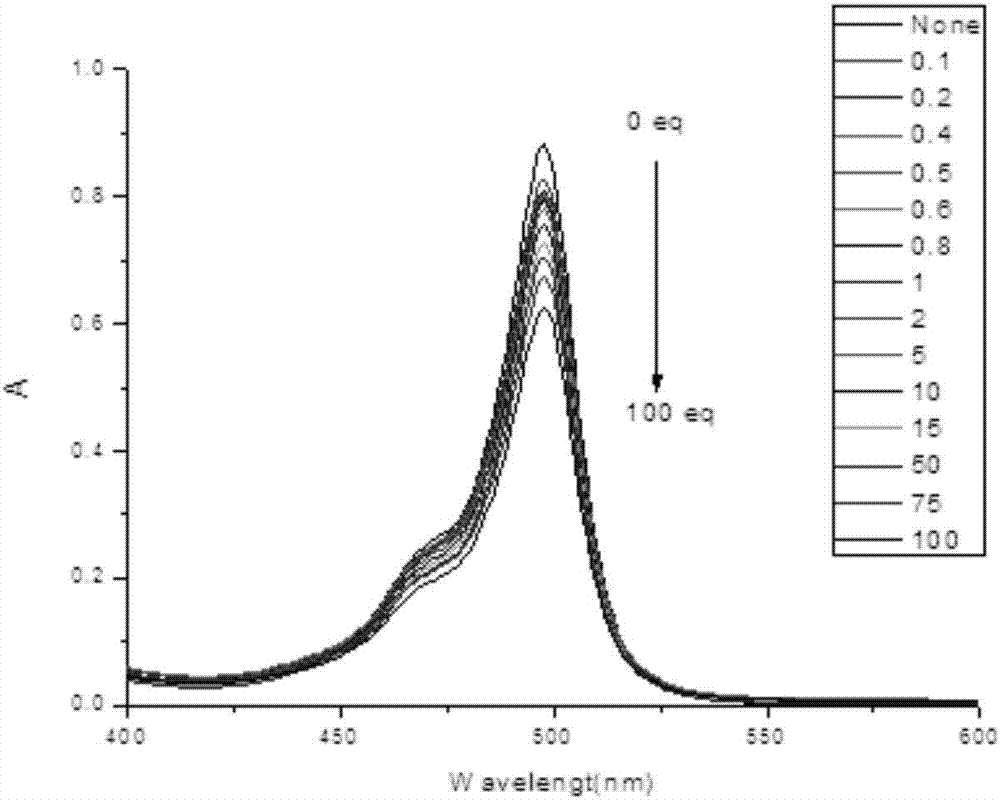
圖2為羧基bodipy在二甲亞碸比水為9:1的溶液中,10-6mol/l羧基bodipy與不同濃度的鋅離子的紫外吸收圖譜,橫坐標為波長、縱坐標為吸光度。鋅離子濃度依次為羧基bodipy濃度的0、0.1、0.2、0.4、0.5、0.6、0.8、1、2、5、10、15、50、75、100倍。圖2表明,羧基bodipy的吸光度隨著鋅離子濃度的升高,成明顯下降趨勢。
圖3為羧基bodipy在二甲亞碸比水為9:1的溶液中,10-6mol/l羧基bodipy與10-4mol/l各離子的螢光發射圖譜,橫坐標為波長、縱坐標為相對螢光強度。螢光強度下降越大,表示羧基bodipy對離子識別響應越高。在測試離子中只對鋅離子有明顯響應,說明羧基bodipy對鋅離子能選擇性識別。
圖4為羧基bodipy在二甲亞碸比水為9:1的溶液中,10-6mol/l羧基bodipy與不同濃度的鋅離子的螢光圖譜,橫坐標為波長、縱坐標為相對螢光強度。鋅離子濃度依次為羧基bodipy濃度的0、0.1、0.2、0.4、0.5、0.6、0.8、1.0、2、5、10、15、50、75、100倍。表示羧基bodipy隨著鋅離子濃度的升高,螢光發射強度逐漸下降。
圖5為羧基bodipy在二甲亞碸比水為9:1的溶液中,10-6mol/l羧基bodipy與10-5mol/l鋅離子和10-5mol/l幹擾離子的螢光差值比值圖,縱坐標為螢光差值的比值,比值越接近1說明幹擾離子的幹擾能力越小。從圖5看出,所有比值均接近1,說明其它離子基本不幹擾本發明螢光探針對鋅離子的高靈敏檢測。
圖6為羧基bodipy的紅外光譜圖,確定了羧基bodipy的各官能團,確定了羧基bodipy的結構。
圖7為羧基bodipy的核磁共振氫譜圖,確定了羧基bodipy的結構。
圖8為羧基bodipy的核磁共振碳譜圖,確定了羧基bodipy的結構。
圖9為羧基bodipy的高分辨質譜圖。
具體實施方式
為了進一步說明本發明,給出以下系列具體實施例,但本發明並不受這些具體實施例的限制,任何了解該領域的技術人員對本發明的些許改動將可以達到類似的結果,這些改動也包含在本發明之中。
實施例1
1、4-(2-羥乙氧基)苯甲醛(以下簡稱化合物1)的合成步驟:
向裝有150ml乙腈的250ml三口燒瓶中,加入12.2g(0.1mol)對羥基苯甲醛和6.9g碳酸鉀(5mmol),攪拌均勻後,再加入9.66g(0.72mol)氯乙醇,電磁攪拌,加熱回流8小時,tlc檢測至原料完全消失,停止反應。冷卻至室溫,分出有機層,將有機層用氫氧化鈉溶液洗至無對羥基苯甲醛為止,分出有機層,硫酸鎂乾燥,過濾,濃縮濾液,得到14.9g化合物1。
2、化合物2的合成步驟:
向裝有50ml甲苯的三口燒瓶中,加入1.0g(2.6mmol)化合物1,攪拌均勻後,再加入1.14g(10mmol)戊二酸酐。在100℃條件下,電磁攪拌,加熱回流8小時,tlc檢測至化合物1完全消失,停止反應。旋幹甲苯,用nahco3水溶液水洗3遍以上,分出有機層,硫酸鎂乾燥,過濾,濃縮濾液。得到1.20g化合物2。
3、羧基bodipy的合成步驟:
在氮氣保護下,向裝有80ml乾燥二氯甲烷的三口燒瓶中加入0.28g(1mmol)的化合物2和0.2ml(2.2mol)2,4-二甲基吡咯,攪拌均勻後,再滴加3-4滴三氟乙酸,室溫下攪拌反應一夜,tlc檢測至原料基本消失;再將0.22g(1mmol)ddq(2,3-二氯-5,6-二氰對苯醌)溶於10ml乾燥二氯甲烷中,並加入上述反應體系中,得到的反應液繼續在室溫下攪拌反應6小時,tlc檢測ddq基本消失,此時加入3ml(22mmol)三乙胺,攪拌半個小時後,緩慢加入3ml(20mmol)三氟化硼乙醚,室溫攪拌5-6個小時,結束反應,用3×30ml蒸餾水洗滌,萃取,硫酸鎂乾燥,過濾,濃縮濾液,經矽膠柱層析分離產物,減壓旋幹後得棗紅色固體0.15g,即產物3,羧基bodipy,產率為30%。其分子式為c26h29bf2n2o5,紅外光譜(kbr),v/cm-1:2923,2847(c-h),1729,1706(o=c),1305(c-n),1245(ar-o-c).核磁氫譜(400mhz,cdcl3)δ7.36(s,1h,oh),7.18(d,j=8.0hz,2h),7.03(d,j=8.0hz,2h),5.99(s,2h),4.50(t,j=6.4hz,2h),4.23(t,j=6.4hz,2h),2.56(s,3h),2.40-2.50(m,4h),1.98-2.02(m,2h),1.42(s,3h).高分辨質譜(m/s):計算值c26h29bf2n2o5498.2139(m)+,測量值498.2144。
實施例2
1、4-(2-羥乙氧基)苯甲醛(以下簡稱化合物1)的合成步驟::
向裝有150ml乙腈的250ml三口燒瓶中,加入0.1mol對羥基苯甲醛和5mmol碳酸鉀,攪拌均勻後,再加入0.1mol氯乙醇,電磁攪拌,加熱回流20小時,tlc檢測至原料完全消失,停止反應。冷卻至室溫,分出有機層,將有機層用氫氧化鈉溶液洗至無對羥基苯甲醛為止,分出有機層,硫酸鎂乾燥,過濾,濃縮濾液,得到化合物1。
2、化合物2的合成步驟:
向裝有50ml甲苯的三口燒瓶中,加入2.6mmol化合物1,攪拌均勻後,再加入2.6mmol戊二酸酐。在100℃條件下,電磁攪拌,加熱回流8小時,tlc檢測至化合物1完全消失,停止反應。旋幹甲苯,用nahco3水溶液水洗3遍以上,分出有機層,硫酸鎂乾燥,過濾,濃縮濾液,得到化合物2。
3、羧基bodipy的合成步驟:
在氮氣保護下,向裝有80ml乾燥二氯甲烷的三口燒瓶中加入1mmol的化合物2和1mol的2,4-二甲基吡咯,攪拌均勻後,再滴加3-4滴三氟乙酸。室溫下攪拌反應3h,tlc檢測至原料基本消失,再將4mmolddq(2,3-二氯-5,6-二氰對苯醌)溶於10ml乾燥二氯甲烷中,並加入上述反應體系中。得到的反應液繼續在室溫下攪拌反應6小時,tlc檢測ddq基本消失,此時加入50mmol三乙胺,攪拌半個小時後,緩慢加入50mmol三氟化硼乙醚,室溫攪拌2小時,結束反應,用3×30ml蒸餾水洗滌,萃取,硫酸鎂乾燥,過濾,濃縮濾液,經矽膠柱層析分離產物,減壓旋幹後得棗紅色固體0.15g,即本發明所述螢光探針羧基bodipy。
實施例3
1、4-(2-羥乙氧基)苯甲醛(以下簡稱化合物1)的合成步驟:
向裝有150ml乙腈的250ml三口燒瓶中,加入0.1mol對羥基苯甲醛和5mmol碳酸鉀,攪拌均勻後,再加入1mol氯乙醇,電磁攪拌,加熱回流20小時,tlc檢測至原料完全消失,停止反應。冷卻至室溫,分出有機層,將有機層用氫氧化鈉溶液洗至無對羥基苯甲醛為止,分出有機層,硫酸鎂乾燥,過濾,濃縮濾液,得到化合物1。
2、化合物2的合成步驟:
向裝有50ml甲苯的三口燒瓶中,加入2.6mmol化合物1,攪拌均勻後,再加入52mmol戊二酸酐。在100℃條件下,電磁攪拌,加熱回流40小時,tlc檢測至化合物1完全消失,停止反應。旋幹甲苯,用nahco3水溶液水洗3遍以上,分出有機層,硫酸鎂乾燥,過濾,濃縮濾液,得到化合物2。
3、羧基bodipy的合成步驟:
在氮氣保護下,向裝有80ml乾燥二氯甲烷的三口燒瓶中加入1mmol的化合物2和5mol的2,4-二甲基吡咯,攪拌均勻後,再滴加3-4滴三氟乙酸。室溫下攪拌反應12h,tlc檢測至原料基本消失,再將0.25mmolddq(2,3-二氯-5,6-二氰對苯醌)溶於10ml乾燥二氯甲烷中,並加入上述反應體系中。得到的反應液繼續在室溫下攪拌反應6小時,tlc檢測ddq基本消失,此時加入10mmol三乙胺,攪拌半個小時後,緩慢加入10mmol三氟化硼乙醚,室溫攪拌12小時,結束反應,用3×30ml蒸餾水洗滌,萃取,硫酸鎂乾燥,過濾,濃縮濾液,經矽膠柱層析分離產物,減壓旋幹後得棗紅色固體0.15g,即本發明所述螢光探針羧基bodipy。
以上所述僅為本發明的實施例,並非因此限制本發明的保護範圍,凡是利用本發明說明書及附圖內容所作的等效流程變換,或直接或間接運用在其他相關的技術領域,均同理包括在本發明的專利保護範圍內。