GO‑PLL‑RGDS/VEGF‑siRNA靶向基因藥物的製備、活性和應用的製作方法
2023-07-25 01:51:21 1
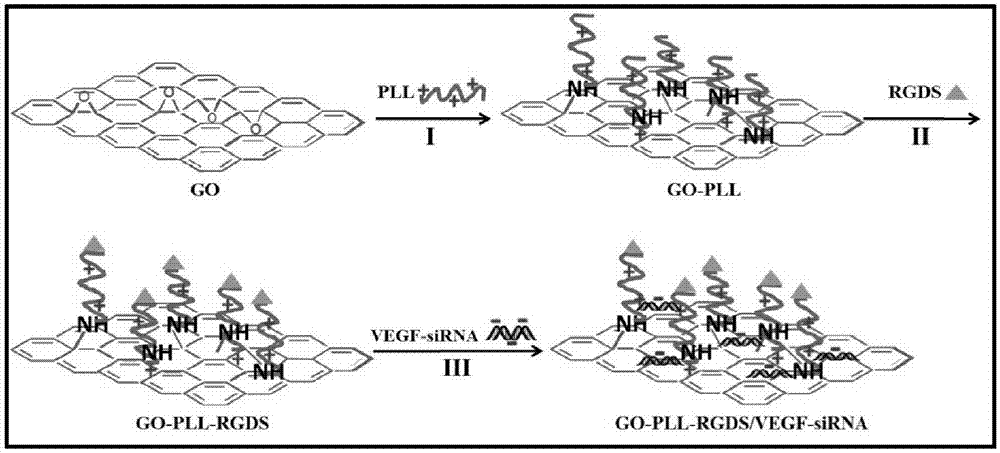
本發明涉及用於遞送vegf-sirna的遞送系統go-pll-rgds/vegf-sirna,涉及它的製備方法,涉及它的納米結構,涉及它的細胞攝取情況,涉及它沉默vegf基因的作用,涉及它下調vegf表達的效率,涉及它抑制腫瘤細胞增殖的作用,並涉及它抑制荷s180肉瘤小鼠腫瘤生長的作用以及遞送系統的腫瘤靶向作用,因而涉及它作為非病毒載體在抗腫瘤基因治療藥物中的應用。屬於生物醫藥學領域。
背景技術:
近年來,研究發現,在腫瘤的基因治療中,非病毒載體比病毒載體安全性高,避免了腺病毒等病毒載體引起免疫原性反應的可能,從而成為基因遞送研究的熱點。目前已報導的非病毒載體主要分為陽離子聚合物類(如聚乙烯亞胺pei)、脂質體類、無機納米材料(如納米金剛石、金納米粒)等,然而大多研究仍存在一些亟需解決的問題。例如,pei的細胞毒性較大;脂質體的負載效率低;無機納米材料大多為疏水性,穩定性以及生物相容性較差。另外,非病毒載體的腫瘤靶向性差也是造成基因遞送效率低、體內毒副作用大的關鍵因素。
石墨烯自發現以來,由於其獨特的單原子層結構特徵和理化性質引起極大關注,除在電學、光學等領域的應用引起廣泛研究外,其越來越多的醫學應用價值被逐漸發掘出來。氧化石墨烯(grapheneoxide,go)是石墨烯的氧化物,是在運用氧化還原法製備石墨烯時得到的前驅體,其表面存在羧基、羥基、環氧基等多種含氧功能團,生物相容性較好並且有利於對其進行功能化修飾。另外,由於其獨特的二維納米結構,氧化石墨烯的比表面積比一般納米載體大,並且上下兩面都可以進行修飾。聚l-賴氨酸(pll)是一種鹼性胺基酸聚合物,由於含有多個游離的氨基,所以在生理條件下發生解離,帶有正電荷。除此之外,pll是一種親水性較強的化合物,在體內可被生物降解,其單體是人體必需胺基酸賴氨酸,所以生物相容性較好。rgd(精氨醯-甘氨醯-天冬氨酸)序列肽是細胞外基質(extracellularmatrix,ecm)中糖蛋白成分纖連蛋白(fibronectin,fn)的保守性序列,對纖連蛋白與細胞表面受體之間的相互作用起到識別作用。rgd序列肽的受體—整合素,是一類膜糖蛋白家族。已有研究表明,在某些實體瘤細胞膜上整合素受體高表達。rgds(精氨醯-甘氨醯-天冬氨醯-絲氨酸)是一種線性rgd序列肽,研究表明可以與腫瘤細胞膜上高表達的整合素受體特異性結合,因此可作為靶向序列對藥物或基因載體進行修飾,實現藥物或基因的靶向遞送。
本發明利用聚l-賴氨酸pll對氧化石墨烯進行正電性和親水性修飾,然後利用靶向序列肽rgds進行靶向性修飾,通過共價鍵將其連接,得到基因vegf-sirna的遞送載體go-pll-rgds。本發明為提高vegf-sirna的負載量,選用比表面積較大的氧化石墨烯作為載體,並通過與pll與靶向性序列肽rgds共價連接製得go-pll-rgds,然後負載vegf-sirna,結果發現1mg的go-pll-rgds可負載100μg的vegf-sirna,基因敷在效率大大提高,並通過藥物分布研究確證了遞送系統的腫瘤靶向性。根據這個發現,發明人提出了本發明。
技術實現要素:
本發明的目的在於提供一種具有抗腫瘤活性的基因遞送系統:go-pll-rgds/vegf-sirna。
本發明所述的基因遞送系統,由抗腫瘤基因治療藥物vegf-sirna和靶向遞送載體go-pll-rgds製備而成。
本發明所述的基因遞送系統,go-pll-rgds與vegf-sirna的質量比為:10:1。
本發明所述的基因遞送系統,為納米結構。
本發明所述的基因遞送系統,靶向遞送載體go-pll-rgds由以下成分製成:go、pll、rgds。
本發明的另一個目的在於提供基因遞送系統的製備方法。
本發明所述的製備方法:將go-pll-rgds與vegf-sirna按照質量比10:1混合,室溫靜置,即得go-pll-rgds/vegf-sirna。
其中,靶向遞送載體go-pll-rgds的製備:氧化石墨烯go通過開環反應、縮合反應依次連接pll(聚l-賴氨酸)和rgds並脫保護得到go-pll-rgds。優選的,本發明所述的基因遞送系統的製備方法,包括以下步驟:
1)製備go-pll
稱取2.0mggo超聲分散在4ml超純水中,用探頭超聲儀即超聲波破碎儀超聲4h(2秒間歇式)。加入koh水溶液(ph9)至8ml。加入10.0mgpll,此時有沉澱物生成,繼續用koh水溶液(ph11)調至ph值為9。將反應瓶置於70℃油浴中攪拌24h。24h後停止加熱,離心(12000g,20min),棄掉上清液,水洗三次,洗掉未反應的pll。凍幹備用。
2)製備boc-arg(tos)-gly-asp(ome)-ser-oh
按照標準方法製備boc-arg(tos)-gly-asp(ome)-ser-obzl。將boc-arg(tos)-gly-asp(ome)-ser-obzl430mg加入20ml甲醇中,加入64.5mg鈀碳,通入氫氣,室溫反應,tlc監測反應。5h後反應完全,濾除鈀碳,旋除甲醇,得到boc-arg(tos)-gly-asp(ome)-ser-oh。
3)製備go-pll-rgds
稱取10mgboc-arg(tos)-gly-asp(ome)-ser-oh,將其溶解在10mldmf中,加入30mgedc·hcl,攪拌活化15min,形成反應液a。將2mggo-pll超聲分散在10mldmf中,然後將其與上述活化的反應液a混合,再加入30mghobt,室溫攪拌。24h後,將反應液離心(12000g,20min),收集沉澱物,用甲醇和超純水洗滌離心三次,除去未結合的boc-arg(tos)-gly-asp(ome)-ser-oh和未反應的edc·hcl和hobt。凍幹得到go-pll-ser-asp(ome)-gly-arg(tos)-boc。將其分散在10ml甲醇溶液中,超聲30min,冰浴條件下,用naoh(2n)溶液調節其ph值等於12,反應6h,待反應結束後,以10000g的轉速離心10min,用超純水漂洗3次,再用無水乙醇漂洗三次,以12000g轉速離心15min,放置通風處自然乾燥。冰浴下,將上述乾燥的產物分散在5ml混合酸液中(tfa:tfoh=4:1,v/v),反應1h。將其轉移至離心管中,離心(7000g,15min)棄掉上清。分別用無水乙醚漂洗和超純水漂洗三次即得到目標產物go-pll-ser-asp-gly-arg(go-pll-rgds)。最後加入5ml超純水,超聲分散,發現產物分散性良好。將分散液裝入透析袋(截留分子量:8000),透析24h除去雜質。將透析純化後的分散液凍幹,測定紅外光譜。
4)製備go-pll-rgds/vegf-sirna
稱取2mggo-pll-rgds,分散在2mldepc水中,超聲40min,分別配製成1000μg/ml的go-pll-rgds儲備液,再將1.0odvegf-sirna(33μg)溶解在250μldepc水中。將go-pll-rgds與vegf-sirna按照一定的n/p比混合,室溫靜置30min,即得go-pll-rgds/vegf-sirna。
本發明的另一個目的在於提供基因遞送系統在製備抑制腫瘤的藥物中的應用。
本發明的另一個目的在於評價基因遞送系統go-pll-rgds/vegf-sirna在基因和蛋白水平下調vegf表達的效率。
本發明的另一個目的在於評價基因遞送系統go-pll-rgds/vegf-sirna抑制腫瘤細胞增殖的作用。
本發明的另一個目的在於評價基因遞送系統go-pll-rgds/vegf-sirna抑制荷s180肉瘤小鼠腫瘤生長的作用。
本發明的另一個目的在於評價基因遞送系統go-pll-rgds/vegf-sirna靶向荷s180肉瘤小鼠腫瘤的靶向性作用。
對說明書中出現的以下詞語作進一步的解釋:
go:grapheneoxide,氧化石墨烯
pll:聚l-賴氨酸
rgds:arg-gly-asp-ser,精氨醯-甘氨醯-天冬氨醯-絲氨酸
說明書中其他位置出現的英文縮寫,請羅列出來作出進一步的解釋:
vegf:血管內皮生長因子
pei:聚乙烯亞胺
tlc:薄層層析法
dmf:n,n-二甲基甲醯胺
edc·hcl:1-(3-二甲基氨基丙基)-3-乙基碳化二亞胺鹽酸鹽
hobt:1-羥基苯並三唑
tfa:三氟乙酸
tfoh:三氟甲磺酸
od值:光密度值
depc:焦碳酸二乙酯
n/p比:氮/磷比,指基因載體與基因的比例
nc:非同源-sirna
dmem:改良杜氏伊格爾培養基
fbs:胎牛血清
fam:羥基螢光素
mtt:3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴鹽
dmso:二甲基亞碸
ns:生理鹽水
cy3:花青染料螢光標記
elisa:酶聯免疫吸附測定
rt-qpcr:實時定量逆轉錄-聚合酶鏈式反應
附圖說明:
圖1.go-pll-rgds/vegf-sirna的製備路線。
註:反應條件:i:koh,70℃.ii:boc-arg(tos)-gly-asp(ome)-ser-oh/edc;naoh/ch3oh;三氟乙酸/三氟甲磺酸.ⅲ:室溫孵育靜置30min.
圖2.瓊脂糖凝膠阻滯實驗結果圖。
圖3.go、go-pll-rgds和go-pll-rgds/vegf-sirna的透射電鏡圖。
註:go(a),go-pll-rgds(b),go-pll-rgds/vegf-sirna(c)的透射電子顯微鏡圖片
圖4.空白水,go、go-pll-rgds和go-pll-rgds/vegf-sirna的原子力顯微鏡。
註:水(a),go(b),go-pll-rgds(c),go-pll-rgds/vegf-sirna(d)的原子力顯微鏡圖片。
圖5.hela細胞對go-pll-rgds/vegf-sirna攝取的結果圖。
a:空白對照組
b:裸fam-vegf-sirna組
c:go-pll-rgds/vegf-sirna組
d:lipo/fam-vegf-sirna組
圖6.go-pll-rgds/vegf-sirna的實時反轉錄定量聚合酶鏈反應的測定結果。(n=3)
註:gpr為go-pll-rgds的指代寫法;nc為非同源陰性對照sirna。
圖7.go-pll-rgds/vegf-sirna的酶聯免疫吸附法測定結果圖。(n=3)
註:gpr為go-pll-rgds的指代寫法;nc為非同源陰性對照sirna。
圖8.mtt法測go-pll-rgds的細胞毒性結果。(n=3)
圖9.go-pll-rgds/vegf-sirna抑制hela細胞增殖作用結果。(n=3)
註:gpr為go-pll-rgds的指代寫法;nc為非同源陰性對照sirna。
圖10.go-pll-rgds/vegf-sirna的體內抗腫瘤活性結果。(n=10)
註:blankcontrol為空白對照生理鹽水組;negativecontrol為vegf-sirna陰性對照組;positivecontrol為阿黴素組。gpr為go-pll-rgds的指代寫法。
給藥劑量:空白對照生理鹽水組為0.2ml/只,陰性對照組vegf-sirna給藥劑量為0.3mg/kg,gpr/vegf-sirna組中go-pll-rgds劑量為3mg/kg,vegf-sirna給藥劑量為0.3mg/kg,陽性對照阿黴素劑量為2μmol/kg。
a為各組腫瘤照片,b為腫瘤重量,c為抑瘤率。
圖11.go-pll-rgds/vegf-sirna的腫瘤靶向作用。
a:空白對照組
b:裸cy3-vegf-sirna組
c:go-pll-rgds/cy3-vegf-sirna組
具體實施方式:
為了進一步闡述本發明,下面給出一系列實施例。這些實施例完全是例證性的,它們僅用來對本發明進行具體描述,不應當理解為對本發明的限制。
實施例1製備go-pll-rgds/vegf-sirna
製備go-pll
稱取2.0mggo超聲分散在4ml超純水中,用探頭超聲儀即超聲波破碎儀超聲4h(2秒間歇式)。加入koh水溶液(ph9)至8ml。加入10.0mgpll,此時有沉澱物生成,繼續用koh水溶液(ph11)調至ph值為9。將反應瓶置於70℃油浴中攪拌24h。24h後停止加熱,離心(12000g,20min),棄掉上清液,水洗三次,洗掉未反應的pll。凍幹備用。測定紅外光譜。傅立葉轉換紅外線光譜(ftir,thermoscientifictmnicolettmistm5,cm-1):3242.74,2935.15,1635.09,1520.51,1340.47,1037.13,601.35。
製備boc-arg(tos)-gly-asp(ome)-ser-oh
按照標準方法製備boc-arg(tos)-gly-asp(ome)-ser-obzl。將boc-arg(tos)-gly-asp(ome)-ser-obzl430mg加入20ml甲醇中,加入64.5mg鈀碳,通入氫氣,室溫反應,tlc監測反應。5h後反應完全,濾除鈀碳,旋除甲醇,得到boc-arg(tos)-gly-asp(ome)-ser-oh。
測定核磁共振,1h-nmr(500mhz,dmso-d6):δ/ppm=δ/ppm=12.656(s,1h),8.169(d,j=7.50hz,1h),8.047(t,j=8.00hz,1h),7.924(d,j=7.50hz,1h),7.630(d,j=8.00hz,2h),7.283(d,j=8.50hz,2h),7.016(m,1h),6.915(d,j=8.00hz,1h),6.614(m,1h),4.725(dd,j1=13.50,j2=7.50hz,1h),4.209(m,1h),3.890(m,1h),3.782(d,j=5.50hz,2h),3.711(dd,j1=11.00hz,j2=5.00hz,1h),3.619(dd,j1=11.00hz,j2=5.00hz,1h),3.595(s,3h),3.308(s,2h),3.029(s,2h),2.746(dd,j1=16.00hz,j2=5.50hz,1h),2.531(dd,j1=16.00hz,j2=8.00hz,1h),2.343(s,3h),1.603(s,1h),1.450(m,2h),1.379(s,9h).
13c-nmr(125mhz,dmso-d6):δ/ppm=172.87,172.15,170.96,170.63,169.18,157.10,155.89,142.26,141.49,129.50,126.04,78.68,61.71,55.33,54.43,51.98,49.55,42.47,36.67,29.63,28.67,21.35.
esi-ms(m/z):[m-1]-:700.47;[2m-1]-:1401.70.
製備go-pll-rgds
稱取10mgboc-arg(tos)-gly-asp(ome)-ser-oh,將其溶解在10mldmf中,加入30mgedc·hcl,攪拌活化15min,形成反應液a。將2mggo-pll超聲分散在10mldmf中,然後將其與上述活化的反應液a混合,再加入30mghobt,室溫攪拌。24h後,將反應液離心(12000g,20min),收集沉澱物,用甲醇和超純水洗滌離心三次,除去未結合的boc-arg(tos)-gly-asp(ome)-ser-oh和未反應的edc·hcl和hobt。凍幹得到go-pll-ser-asp(ome)-gly-arg(tos)-boc。將其分散在10ml甲醇溶液中,超聲30min,冰浴條件下,用naoh(2n)溶液調節其ph值等於12,反應6h,待反應結束後,以10000g的轉速離心10min,用超純水漂洗3次,再用無水乙醇漂洗三次,以12000g轉速離心15min,放置通風處自然乾燥。冰浴下,將上述乾燥的產物分散在5ml混合酸液中(tfa:tfoh=4:1,v/v),反應1h。將其轉移至離心管中,離心(7000g,15min)棄掉上清。分別用無水乙醚漂和超純水漂洗三次即得到目標產物go-pll-ser-asp-gly-arg(go-pll-rgds)。最後加入5ml超純水,超聲分散,發現產物分散性良好。將分散液裝入透析袋(截留分子量:8000),透析24h除去雜質。將透析純化後的分散液凍幹,測定紅外光譜。傅立葉轉換紅外線光譜(ftir,thermoscientifictmnicolettmistm5,cm-1):3253.22,2922.98,1643.00,1518.86,1056.93,666.97。
製備go-pll-rgds/vegf-sirna
稱取2mggo-pll-rgds,分散在2mldepc水中,超聲40min,分別配製成1000μg/ml的go-pll-rgds儲備液,再將1.0odvegf-sirna(33μg)溶解在250μldepc水中。將go-pll-rgds與vegf-sirna按照一定的n/p比混合,室溫靜置30min,即可得到轉染複合物go-pll-rgds/vegf-sirna。
測定go-pll-rgds/vegf-sirna中vegf-sirna的負載量
配製樣品用rna稀釋緩衝液配製go-pll-rgds:vegf-sirna(質量比)為2:1,8:1,10:1,20:1,30:1,40:1,50:1的混合懸浮液100μl,搖勻,室溫靜置30min。
實驗步驟稱取0.5g瓊脂糖於100ml燒杯中,加入5mltbe(10×)和45ml去離子水,置於微波爐內中高火加熱2min將瓊脂糖溶化。稍冷卻後加入10μl溴化乙錠溶液(10mg/ml),攪勻,倒入模具中(事先洗淨並用無水乙醇潤洗晾乾),插入梳子,凝固後備用。取30mltbe(10×)和270ml超純水,混合均勻後倒入電泳槽中作為電泳液。取出凝固後的瓊脂糖凝膠,置於電泳槽中,每孔加樣20μl。打開電源,將電壓設置成120v,時間設置為20min。從電泳槽中取出瓊脂糖凝膠,置於凝膠成像儀觀察並拍照。
實驗結果如圖2所示,不同質量比的go-pll-rgds/vegf-sirna的螢光條帶均比游離sirna組(對照)弱,說明vegf-sirna被不同程度地阻滯在上樣孔裡,表明vegf-sirna成功被部分或完全穩定負載在go-pll-rgds表面。當質量比為10:1時,螢光條帶完全消失,說明此時vegf-sirna被完全阻滯在上樣孔裡,即vegf-sirna被完全負載在go-pll-rgds表面。所以,1mggo-pll-rgds可以負載100μgvegf-sirna。
實施例2測定go-pll-rgds和go-pll-rgds/vegf-sirna的納米結構
實驗方法取100μl濃度為1000μg/ml的go、go-pll-rgds水分散液用純水稀釋到2ml。按照質量比10:1配製go-pll-rgds/vegf-sirna水分散液,即go-pll-rgds的濃度為50μg/ml,vegf-sirna的濃度為5μg/ml。各取20μl滴加到碳支持膜(銅網)上,置於37℃烘箱中乾燥24h。將各樣品置於透射電鏡(tem,jem-1230,jeol,日本)下觀察並獲取照片。再各取20μl滴加到雲母片上,置於室溫自然風乾。將各樣品置於原子力顯微鏡(afm,veecoinstruments,inc,plainview,usa)下觀察並獲取照片。以水為空白背景。
實驗結果從圖3中的透射電鏡圖像我們可以看出go有典型的褶皺片狀納米結構,粒徑大小不均,在100-350nm範圍內。而go-pll-rgds的粒徑大小明顯較小,在60-180nm之間,。這是由於親水性化合物pll和rgds的修飾,使go-pll-rgds的水分散性更好,不易發生聚集。而負載了vegf-sirna的go-pll-rgds/vegf-sirna的粒徑增加,部分達到200nm以上。另外,圖4中的原子力顯微鏡圖像顯示,go-pll-rgds的厚度由未經修飾的1.7nm左右(go)增加到3.0nm。與go-pll-rgds相比,go-pll-rgds/vegf-sirna的粒徑和厚度均有增加,厚度增加到4.4nm左右。
實施例3測定go-pll-rgds和go-pll-rgds/vegf-sirna的zeta電位和水合粒徑
實驗方法配製100μg/mlgo和go-pll-rgds的水分散液備用。配製100μg/ml(即go-pll-rgds的濃度為100μg/ml,vegf-sirna的濃度為10μg/ml)的go-pll-rgds/vegf-sirna水分散液。用雷射粒度分析儀測定樣品的粒徑分布與zeta電位。
實驗結果go、go-pll-rgds和go-pll-rgds/vegf-sirna的zeta電位值分別是-40.59±2.23mv、25.87±0.47mv、15.36±2.62mv。go、go-pll-rgds和go-pll-rgds/vegf-sirna的水合粒徑分別為275.8±2.7nm、162.9±4.5nm、196.6±2.7nm。zeta電位值由負值變為正值,也進一步說明go-pll-rgds的成功修飾。在負載vegf-sirna之後,複合物go-pll-rgds/vegf-sirna仍表現出一定的正電性,這對細胞攝取有重要意義。
實施例4雷射共聚焦顯微鏡考察hela細胞對go-pll-rgds/vegf-sirna的細胞攝取情況
實驗方法
1)藥物配製:將go-pll-rgds超聲分散在depc水中,配成濃度為1000μg/ml的儲備液。將其放置在無菌樣品瓶中(厚度不超過1cm),紫外照射24h。將1.0od(2.5nmol,33μg)fam-vegf-sirna溶解在250μl的depc水中,配成10nmol/ml的儲備液。lipo(lipornaimax)的使用濃度按照說明書推薦量。按照以下各組分比例分別配成100nmol/l(按sirna的濃度計)的裸fam-vegf-sirna、go-pll-rgds/fam-vegf-sirna、lipo/fam-vegf-sirna。
1.裸fam-vegf-sirna(陰性對照組):取24μlfam-vegf-sirna儲備液加入到2376μl無血清培養基中混勻備用。
2.go-pll-rgds/fam-vegf-sirna(實驗組):取24μlfam-vegf-sirna和34μlgo-pll-rgds儲備液加入到2342μl無血清培養基中混勻,室溫靜置30min備用。
3.lipo/fam-vegf-sirna(陽性對照組):取24μlfam-vegf-sirna和20μllipo(lipornaimax)儲備液加入到2356μl無血清培養基中混勻,室溫靜置30min備用。
2)細胞復甦及培養:
1.打開水浴鍋,預熱(37℃)。
2.從液氮罐中取出凍存的hela細胞,迅速放入37℃水浴中,1min內溶解。
3.在超淨臺中把溶解的細胞快速移入離心管中,加入2ml的完全培養基。離心(1000g,5min)。
4.棄上清,加入1ml培養基,吹打均勻,移入25cm2培養瓶中,補加4ml培養基,37℃、5%co2的培養箱中培養。
3)細胞傳代:棄掉原培養基,加入2ml的pbs小心吹洗2次,再加入1ml胰酶,吹打均勻,放入培養箱,4min後,在倒置顯微鏡下觀察,細胞回縮成球形懸浮起來。加入3ml完全培養基,終止消化。小心吹打細胞,並將其轉入離心管中,離心(1000g,5min),小心傾倒上清液,加入4ml完全培養基吹打均勻。各吸取2ml細胞懸液於25cm2培養瓶中。再各自補加3ml培養液,吹打均勻,放入培養箱中,37℃、5%co2培養。
4)雷射共聚焦顯微鏡測定:將3×105個(2.0×105/ml)hela細胞種於內徑10mm的雷射共聚焦小皿中,37℃、5%co2培養24h,待細胞貼壁後換成配製好的含藥培養基(不含fbs)。分別設空白對照組(加入等體積的空白培養基)、陽性對照組、陰性對照組、實驗組。轉染4h後,棄掉含藥培養基,用pbs洗三次,每皿加入hoechst33342工作液1ml,孵育15min後棄掉,用pbs洗三遍,最後在每皿中預留少量pbs防止乾片。置於雷射共聚焦顯微鏡下觀察並拍照。
實驗結果fam是一種羧基化的螢光素類標記試劑,激發波長492nm,發射波長518nm。如圖5所示,a組是空白對照組,hoechst33342將細胞核染成藍色。b組鏡下只能觀察到藍色的細胞核,可知,游離的fam-vegf-sirna不能被細胞攝取。c組中,在細胞核外能觀察到綠色螢光,在merge通道下可以觀察到綠色螢光集中分布在胞質中,可知,fam-vegf-sirna被go-pll-rgds成功遞送進入細胞。d組作為陽性對照,也可以在細胞核周圍觀察到綠色螢光。因此,從以上結果可以得出以下結論:轉染複合物go-pll-rgds/fam-vegf-sirna成功被細胞攝取,並定位於細胞質內。
實施例5實時反轉錄定量聚合酶鏈反應評價go-pll-rgds/vegf-sirna沉默vegf編碼基因的作用
實驗方法
1)藥物配製:將go-pll-rgds超聲分散在depc水中,配成濃度為1000μg/ml(儲備液1)的儲備液。將其分別放置在無菌樣品瓶中(厚度不超過1cm),紫外照射24h備用。將1.0od(2.5nmol,33μg)homo-vegf-sirna溶解在250μl的depc水中,配成10nmol/ml的儲備液。1.0od(2.5nmol,33μg)非同源-sirna(nc)溶解在250μl的depc水中,配成10nmol/ml的儲備液。lipo(lipornaimax)的使用濃度按照說明書推薦量。
按下表配製含藥培養基各6ml(濃度為100nm):
表1.藥物配製比例
2)細胞轉染:取對數期hela細胞,計數,調整細胞濃度至1.6×105個/ml(所用培養基為不含抗生素但含10%fbs的dmem培養基)。每孔吸取2ml細胞懸液接種到六孔板中,使每孔細胞個數為3.2×105個,37℃、5%co2培養。12h後待細胞貼壁後,將培養基小心吸出。每孔加入配製的含藥培養基各2ml,每組設3個復孔,空白對照組加入等體積的dmem。37℃、5%co2條件下繼續培養6h。6h後,小心吸出含藥培養基,換成完全培養基,繼續培養42h。
在進行細胞轉染時,我們發現,如果給予轉染試劑lipo(lipornaimax)前的細胞用含雙抗的完全培養基培養,當加入轉染試劑lipo後發現細胞在30min內形狀由梭形變成圓形,漂浮在培養基中。而go-pll-rgds/vegf-sirna則無此現象。之後,用不含雙抗的培養基培養至貼壁,然後給予轉染試劑進行轉染,此現象消失。
3)細胞全rna的提取:操作前用rna酶清除劑擦拭工作檯面,使用無酶離心管和槍頭。
1.取轉染結束的六孔板,棄去培養基,每孔加入1mltrizol裂解液(trizol裂解液的用量根據孔面積計算,10cm2加1mltrizol裂解液),用移液槍吹打均勻,3min後轉移到離心管中,靜置5min,使得核酸蛋白複合物完全分離。
2.每管加入0.2ml氯仿,劇烈震蕩15s,室溫靜置3min,離心(4℃,12000g,15min)。
3.離心後混合液分成三層,下面為紅色的苯酚-氯仿層,上面為水相,rna存在水相中,約佔總體積的50%。
4.沉澱rna:將上層水相緩慢小心地轉移到另一個離心管中,加入0.5ml異丙醇,混勻,室溫靜置10min,然後離心(4℃,12000g,15min),可見白色沉澱。
5.洗滌rna:棄上清,加入1ml配置好的75%乙醇(v醇/vdepc水=3:1)。洗滌rna沉澱,震蕩搖勻,離心(4℃,12000g,15min),重複洗滌三次。棄上清,晾乾(注意:無需完全乾燥,否則極大降低它的溶解度)。
6.rna再溶解:用depc水(每管60μl)重懸rna,吹打幾次,用低功率超聲使rna完全溶解,55℃水浴孵育10min,再冷卻至室溫,分裝後-80℃保存。
7.純度及濃度測定:用depc水校正nanodrop-1000後,用2μlrna水溶液滴在測量臂上,選擇核酸rna-measure進行測量od260/od280值和濃度值。
4)反轉錄:
1.樣品準備:根據測定的rna濃度,計算出所需的rna體積(樣品需2μg),配置樣品。
2.每個反應孔加10μl2×rtbuffer,1μlrtenzymemix和相應體積的rna溶液,depc水補足20μl,加入到microamptmoptical96-wellreactionplate中,用封口膜封口,4℃,1000g,離心3min,除去氣泡。
3.將microamptmoptical96-wellreactionplate放入realtimepcrsystem7500中,設置條件,94℃5min,94℃30s,55℃30s,72℃25s,72℃7min,4℃∞。共30個循環。獲得cdna,測其濃度。
5)聚合酶鏈反應:
1.cdna的上樣量為50ng,根據其濃度計算需加入的各組cdna樣品的體積。按照以下各組分體積配製混合液:每個反應孔混合液體積為50μl,包括25μlgeneexpressionmastermix、2.5μl探針標記的引物(vegf或gapdh)、相應體積的cdna樣品和depc水。
2.將配好的混合液加入microamptmoptical96-wellreactionplate中,每孔50μl,離心(4℃,1000g,3min),使混合液沉於反應孔底部並排出氣泡,
3.將96孔板放入real-timepcrsystem7500中,設置條件,hold:50℃2min,95℃10min,cycle(40cycles),95℃15s,60℃1min。開始擴增。
4.數據處理。以gapdh為內參,運用2-△△ct法相對定量各mrna的量。上述實驗重複三次。
實驗結果
表2.在go-pll-rgds/vegf-sirna作用下hela細胞的vegf-mrna的相對量(均值±sd,n=3)
從圖6可以看出:陰性對照組游離的vegf-sirna和go-pll-rgds/非同源sirna的mrna相對表達量與空白對照組比較,無顯著性差異(p>0.05),即兩者均不能有效下調mrna的表達。而實驗組go-pll-rgds/vegf-sirna的mrna的相對表達量與空白對照組比較有顯著性差異(p0.05),即兩者均不能有效下調vegf的表達。而實驗組go-pll-rgds/vegf-sirna的vegf的相對表達量與空白對照組比較有顯著性差異(p<0.01),go-pll-rgds/vegf-sirna能夠有效下調蛋白的表達,下調率為51.71±4.54%。
實施例7評價go-pll-rgds/vegf-sirna的細胞毒性和抑制腫瘤細胞增殖的作用
實驗方法
1)試劑配製
完全培養基:dmem培養基(450ml)中加入滅活的胎牛血清50ml(10%),青黴素100u/ml,鏈黴素100μg/ml。
無血清培養基:dmem培養基。
胎牛血清的滅活:將成品胎牛血清在56℃水浴中孵育30min,分裝為50ml/份,-20℃保存。
pbs:將pbs粉末按說明用超純水配製成ph7.4,濃度為0.01m的pbs緩衝液,高壓滅菌。4℃保存。
胰酶:將成品胰酶分裝10ml/份,4℃保存。
mtt工作液:稱取50mgmtt粉末,將其溶解在10ml的pbs緩衝液中,超聲使其充分溶解,配製成5mg/ml的工作液,用0.22μm過濾膜過濾除菌。
2)藥物配製
將中間產物go-pll超聲分散在depc水中,配成濃度為1000μg/ml的儲備液1。
將go-pll-rgds超聲分散在depc水中,配成濃度為1000μg/ml(儲備液2)和500μg/ml(儲備液3)的儲備液。將其分別放置在無菌樣品瓶中(厚度不超過1cm),紫外照射24h備用。將1.0od(2.5nmol,33μg)homo-vegf-sirna溶解在500μldepc水中,配成5nmol/ml的儲備液。1.0od(2.5nmol,33μg)非同源-sirna(nc)溶解在500μldepc水中,配成5nmol/ml的儲備液4。lipo(lipornaimax)的使用濃度按照說明書推薦量。
1.go-pll-rgds的細胞毒性評價
go-pll和go-pll-rgds:將go-pll和go-pll-rgds儲備液分別稀釋成濃度分別為50,100,200,300,400,500,750μg/ml的go-pll母液和go-pll-rgds母液。
2.go-pll-rgds/vegf-sirna的抑制hela細胞增殖的作用
空白背景組:只加入空白培養基,沒有細胞。
空白對照組:不進行處理,只加入相同體積的培養基的細胞。
陰性對照組:go-pll-rgds、vegf-sirna、go-pll-rgds/非同源-sirna、lipo/非同源-sirna
陽性對照組:lipo/vegf-sirna
實驗組:go-pll-rgds/vegf-sirna、go-pll-rgds、vegf-sirna、go-pll-rgds/非同源-sirna、lipo/非同源-sirna、lipo/vegf-sirna和go-pll-rgds/vegf-sirna的濃度均為10nm,40nm,80nm,120nm。
3)mtt法評價go-pll-rgds的細胞毒性
取對數期hela細胞,計數,調整細胞濃度至4×104個/ml。96孔板用pbs液封,每孔取100μl細胞懸液種板,37℃、5%co2培養。12h後待細胞貼壁後,給藥。每孔加入上述配製好的go-pll母液或go-pll-rgds母液25μl,使加入的go-pll-rgds或go-pll的終濃度是10,20,40,60,80,100,150μg/ml,每個濃度設6個復孔,空白背景組和空白對照對照組補加25μldepc水。37℃、5%co2條件下繼續培養48h。48h後,每孔加25μlmtt工作液,左右前後搖晃均勻,37℃孵育4h。4h後4000g離心10分鐘。小心吸出每孔培養基。每孔加入dmso150μl,用微型振蕩器震蕩(500r,10min)。在酶標儀上,以檢測波長為570nm測定吸光度(od)值,計算細胞存活率。細胞存活率(%)=(實驗組od值-空白組od值)/(對照組od值-空白組od值)×100%。上述實驗重複三次。
4)go-pll-rgds/vegf-sirna抑制hela細胞增殖的作用
取對數期hela細胞,計數,用完全培養基調整細胞濃度至4×104個/ml。96孔板用pbs液封,每孔取100μl細胞懸液種板,37℃、5%co2培養。12h後,待細胞貼壁,小心吸出完全培養基,每孔加入100μl無血清培養基。然後,每孔分別加入25μl配製不同濃度的的go-pll-rgds、vegf-sirna、go-pll-rgds/非同源-sirna、lipo/非同源-sirna、lipo/vegf-sirna和go-pll-rgds/vegf-sirna進行轉染,各組終濃度分別為10,40,80,120nm(按sirna計)。轉染6h後,各孔換成等體積完全培養基,繼續培養42h。每孔加25μl的mtt工作液,左右前後搖晃均勻,37℃孵育4h。4h後4000g離心10分鐘。小心吸出每孔培養基。每孔加入dmso150μl,用微型振蕩器震蕩500r,15min。在酶標儀上,以檢測波長為570nm測定吸光度(od)值,計算細胞存活率。上述實驗重複三次。
實驗結果
1.go-pll-rgds的細胞毒性評價
理想的基因載體不僅要有效,同樣重要的是低毒性。通過mtt法對go-pll-rgds和中間物go-pll的細胞毒性進行評價。結果如圖8所示,當go-pll-rgds的濃度增加至150μg/ml時,其細胞存活率扔在85%以上,為表現出明顯的細胞毒性。而同濃度的go-pll處理過的細胞,存活率比go-pll-rgds組低,並且當濃度增加至60,80,100,150μg/ml時,二者之間有顯著性差異(p<0.05),即go-pll的細胞毒性大於go-pll-rgds。有文獻報導,正電性的納米載體可以與細胞膜表面負電性的糖蛋白相互作用,從而表現出細胞毒性;並且通過修飾中和納米載體的正電性,可以減小其細胞毒性。在上述章節中,我們已經測得go-pll-rgds的zeta電位值(25.87±0.47mv)明顯小於go-pll的zeta電位值(40.20±4.89mv),即經過rgds的修飾,使pll上的氨基減少,中和了go-pll上部分的正電荷。
2.go-pll-rgds/vegf-sirna的抑制hela細胞增殖的作用
從圖9可以得出:
1.不同濃度的vegf-sirna與空白對照組相比,細胞存活率未表現出顯著性差異,即vegf-sirna對細胞增殖無抑制作用。
2.不同濃度的go-pll-rgds、go-pll-rgds/非同源-sirna和lipo(lipornaimax)/非同源-sirna與空白對照比較,雖然表現出差異(p<0.05),但是細胞存活率均在80%以上,對細胞增殖無明顯抑制作用。
3.陽性對照組lipo(lipornaimax)/vegf-sirna與空白對照組相比,有顯著性差異(p<0.01),表現出明顯的抑制細胞增殖的作用。驗證了實驗方法的正確性。
4.10nm的go-pll-rgds/vegf-sirna與空白對照組比較,細胞存活率表現出顯著性差異(p0.05)。go-pll-rgds/vegf-sirna組的平均瘤重與空白對照生理鹽水組和陰性對照游離vegf-sirna組比較,有顯著性差異(p<0.01),抑瘤率為51.74%,其抑瘤率略小於化療藥dox(56.23%)。上述結果說明游離的vegf-sirna不能有效抑制腫瘤的生長,而go-pll-rgds/vegf-sirna則可有效抑制腫瘤生長,表現出體內抗腫瘤活性。
2)各組荷瘤小鼠血清中vegf的表達水平
實驗方法取上述獲得的小鼠血清樣品,室溫融化,按mus-vegf-elisakit說明描繪標準曲線並測定血清中的vegf蛋白含量。
實驗結果按照說明配製一系列濃度的標準蛋白液,測定其吸收值,繪製標準曲線如下:回歸方程為y=0.0096x+0.1278(r2=0.999)。各組荷瘤小鼠血清中vegf的絕對含量結果如下表所示:go-pll-rgds/vegf-sirna組的小鼠血清中的vegf含量為61.44±2.62pg/ml,明顯低於空白對照組和游離的vegf-sirna組。結果說明,go-pll-rgds/vegf-sirna可以有效下調小鼠體內的vegf蛋白的表達量,即有效使vegf-sirna發揮基因沉默作用。這與體外結果與討論相吻合。
表1.各組小鼠血清中vegf含量(均值±sd,pg/ml)
實施例9評價go-pll-rgds/vegf-sirna的腫瘤靶向性作用—螢光成像法測定go-pll-rgds/vegf-sirna的組織分布
實驗方法:
1.荷s180肉瘤小鼠模型的建立
將0.2ml濃度為1×107個/ml的s180細胞懸液接種於每隻小鼠左腋皮下,建立荷s180肉瘤小鼠模型。小鼠飼養環境整潔衛生,5隻一籠,溫度為18-22℃,溼度為50-60%,飲食充足且自由。
2.給藥
隨機將建模成功的小鼠分為3組,每組3隻。分別按照以下劑量尾靜脈注射給藥,給藥一次。
生理鹽水組:0.2ml/只
cy3-vegf-sirna組:0.3mg/kg
go-pll-rgds/cy3-vegf-sirna組:0.3mg/kg(按cy3-vegf-sirna計);3.0mg/kg(按go-pll-rgds計),配製方法如2.1.3所述。
3.螢光成像觀察
給藥24h後,將小鼠處死,取出各臟器,注意避光。置於螢光成像儀下觀察並定量計算各臟器的螢光強度值。激發波長為550nm,發射波長為570nm。註:12h時先用6%水合氯醛0.2ml/只腹腔注射將小鼠麻醉,然後活體成像,觀察螢光。結果由於體毛影響造成無法成像,之後將其腹部的體毛剃掉,置於螢光成像儀下,結果仍看不到螢光圖像。分析原因:可能由於cy3的螢光強度較弱,小鼠皮膚及組織較厚。所以採取處死小鼠後直接取出臟器後觀察這一方法。註:cy3螢光染料是一種常用於生物分子標記的水溶性染料。
實驗結果如圖11a所示,生理鹽水組的各器官觀察不到螢光,游離的cy3-vegf-sirna組中,在腫瘤、肝、肺、腎以及腦中均可觀察到較弱的螢光,並且腫瘤部位的螢光強度並不比其他臟器的螢光強度強。這是由於游離的cy3-vegf-sirna容易被酶解失活,並較快地排出體外。而go-pll-rgds/cy3-vegf-sirna組中,腫瘤部位的螢光強度明顯強於其他臟器,並且,在腦中未觀察到螢光。另外,腎臟的螢光強度較其他器官的螢光強度強,go-pll-rgds/cy3-vegf-sirna可能通過腎臟排洩,從而在腎臟有蓄積。肺部的螢光面積較游離的cy3-vegf-sirna組肺部螢光面積大,與部分氧化石墨烯會沉積在肺部有關。b中,將螢光強度值量化,可以得出同樣的結果。go-pll-rgds/cy3-vegf-sirna在腫瘤部位的螢光強度值是腎臟的2.34倍。綜上說明給藥24h後,go-pll-rgds/cy3-vegf-sirna大部分在腫瘤組織分布,其次是在腎臟分布,可能與代謝排洩途徑有關;另外在肝臟和肺部也有少量分布。這一結果證明了go-pll-rgds/cy3-vegf-sirna具有腫瘤靶向性。