一種超聲刺激響應性聚醯胺囊泡的製備方法與流程
2023-06-18 06:23:01 4
本發明涉及聚合物材料領域,具體涉及一種超聲刺激響應性聚醯胺囊泡的製備方法。
背景技術:
囊泡是廣泛存在於生命系統中的結構,在基因藥物運輸等領域中有很廣泛的應用,因而已經引起了科研工作者極大的研究興趣。一般來說,囊泡可以由脂質體分子、表面活性劑、嵌段共聚物的自組裝得到。其中,毫無疑問,聚合物囊泡位於納米科技革命的最前沿,這是由於聚合物囊泡在藥物封裝、藥物運輸、納米反應器和微米或納米材料模板等領域擁有無限可能的應用潛力。超聲是一種對人體無害的物理信號,因此超聲刺激響應性聚合物囊泡在生物體內藥物運輸和緩釋具有非常大的前景。然而,目前聚合物囊泡大多數都是由二嵌段共聚物,三嵌段共聚物、樹枝狀聚合物和超支化聚合物的自組裝得到的,而關於利用多嵌段的縮聚物則很難製備出囊泡,並且目前聚合物囊泡很難具備超聲響應性能。
技術實現要素:
為解決上述現有技術中存在的問題,本發明提供一種超聲刺激響應性聚醯胺囊泡的製備方法。
本發明的超聲刺激響應性聚醯胺囊泡的製備方法,包括以下步驟:
(1)向反應球瓶中加入乾燥劑、溶劑,緩慢加熱至40-70℃,攪拌使之完全溶解,隨後再向溶液中加入含磺酸基的芳香族二元羧酸和穩定劑,待其完全溶解後,再加入稍過量的二元胺,隨後關閉控溫儀,待溶液冷卻後,再抽排三次,用惰性氣體置換出體系中的空氣,在保持惰性氣氛下,進行反應,得到親水聚醯胺嵌段;以脂肪族二元羧酸代替含磺酸基的芳香族二元羧酸,以同樣的方法得到疏水聚醯胺鏈段;
(2)迅速將質量比為1:4-3:5的親水聚醯胺鏈段和疏水聚醯胺鏈段混合加入到反應球瓶中,同時補加乾燥劑補償整個反應體系的損失,在惰性氣氛下,繼續反應,得到共聚醯胺溶液,用甲醇使其沉澱,靜置後,過濾、洗滌,收集到多嵌段共聚醯胺,進一步在90-105℃下真空乾燥;所述真空乾燥的時間為20-30小時,優選24小時。
(3)將多嵌段共聚醯胺溶於去離子水中製得多嵌段共聚醯胺溶液,在40-60℃下加熱攪拌,得到超聲刺激響應性聚醯胺囊泡;所述加熱攪拌優選12小時。
根據如上所述的製備方法,在步驟(1)及步驟(2)中,所述乾燥劑為氯化鈣、硫酸鈣、硫酸銅中的一種或幾種。
根據如上所述的製備方法,在步驟(1)及步驟(2)中,所述反應的條件是加熱升溫至95℃-110℃、反應2-20小時。其中,反應溫度更為優選95℃、100℃、105℃或110℃,反應時間更為優選2小時、3小時、4小時、5小時、6小時、7小時、10小時或20小時。
另外,在步驟(3)中,親水聚醯胺鏈段與疏水聚醯胺鏈段的質量比為1:4-3:5,該質量比也可優選1:4、2:5或3:5。
根據如上所述的製備方法,在步驟(1)及步驟(2)中,所述溶劑為n-甲基吡咯烷酮和吡啶的混合溶液,其中n-甲基吡咯烷酮與吡啶的體積比為5-9:1-4,其中更為優選例如5:1、6:2、7:1、8:3或9:4。
根據如上所述的製備方法,在步驟(1)及步驟(2)中,所述穩定劑為磷酸三苯酯、磷酸三甲酯或亞磷酸酯中的一種或幾種。
根據如上所述的製備方法,在步驟(1)及步驟(2)中,所述二元胺為乙二胺、丁二胺、己二胺或葵二胺中的一種或幾種。
根據如上所述的製備方法,在步驟(1)中,所述含磺酸基的芳香族二元羧酸至少為間苯二甲酸-5-磺酸鈉、對苯二甲酸-5-磺酸鈉中的一種或幾種
根據如上所述的製備方法,在步驟(1)中,所述的脂肪族二元酸為丁二酸、己二酸、壬二酸或癸二酸中的一種或幾種。
所述的製備方法,步驟(1)中親水聚醯胺鏈段製備中各成分用量的質量比為60-100份混合溶劑:20-30份含磺酸基的芳香族二元羧酸:6-18份二元胺:60-100份穩定劑:1-6份乾燥劑;步驟(1)中疏水聚醯胺鏈段製備中各成分用量的質量比為60-100份混合溶劑:12-20份脂肪族二元酸:6-18份二元胺:60-100份穩定劑:1-6份乾燥劑;
根據如上所述的製備方法,在步驟(3)中,所述反應的反應時間為24-96小時、反應溫度為100-130℃,其中反應時間優選為24小時、48小時、72小時或96小時;反應溫度優選100℃、105℃、120℃或130℃。
另外,在步驟(3)中,真空乾燥溫度為90-105℃,其中優選90℃、95℃、100℃或105℃。
根據如上所述的製備方法,在步驟(4)中,多嵌段共聚醯胺溶液的濃度為0.5mg/ml-10mg/ml。其中,優選0.5mg/ml、1mg/ml或2mg/ml。
另外,在步驟(4)中,攪拌溫度為40-60℃,可優選為40℃、50℃或60℃。
由本發明所製備的聚醯胺囊泡具有超聲響應性,在超聲的作用下囊泡會發生破裂。本發明所涉及的製備方法簡單,囊泡性能穩定。
選用不溶於水的尼羅紅分子作為模型藥物分子,對囊泡水溶液而言,如果包裹在囊泡疏水壁中的尼羅紅分子被釋放到水溶液中,由於尼羅紅分子不溶於水,會聚集沉澱下來,從而導致囊泡水溶液中尼羅紅染料螢光強度的降低,因此可以通過檢測囊泡水溶液的螢光光譜,來確定mbcpa囊泡的藥物釋放能力,並且可以通過與hbpo-star-peo囊泡的釋放進行比較來驗證本發明超聲響應的獨特性。
通過對比室溫下、50℃、50℃超聲作用下囊泡的螢光發射光譜來驗證本發明中囊泡的穩定性。
附圖說明
圖1是實施例1製得的mbcpa的核磁圖譜。
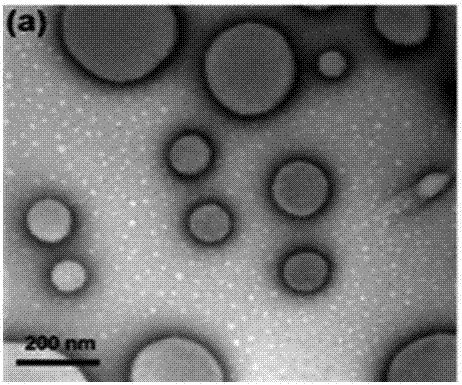
圖2是實施例1製得的囊泡的透射電鏡圖。
圖3是實施例1中的在不同超聲作用時間後囊泡溶液中尼羅紅紫外吸收強度的測試結果。
圖4是超聲作用後囊泡溶液圖。
圖5是在超聲作用下包裹有尼羅紅染料分子的實施例1製得的囊泡或hbpo-star-peo囊泡水溶液的螢光發射光譜歸一化曲線。
圖6是實施例1製得的囊泡在室溫下、50℃、50℃超聲作用下囊泡的螢光發射光譜歸一化曲線。
具體實施方式
下面結合實施例對本發明進行詳述,但本發明並不限定於這些實施例。
下述實施例中所使用的實驗方法如無特殊說明,均為本領域技術人員所知的常規方法。下述實施例中所用的材料、試劑等,如無特殊說明,均可從商業途徑常規獲得。
一、超聲刺激響應性聚醯胺囊泡的製備
實施例1
1)在50ml的反應球瓶中加入0.1g的無水氯化鈣、5.0ml的n-甲基吡咯烷酮與1ml的吡啶的混合溶液,緩慢加熱至70℃,攪拌使之完全溶解。隨後再向溶液中加入2.68g間苯二甲酸-5-磺酸鈉(5-ssipa)和5.2ml亞磷酸三苯酯,待其完全溶解後,再加入稍過量的1.2g己二胺。隨後關閉控溫儀,待溶液冷卻後,再抽排三次,用氮氣置換出體系中的空氣。在保持體系惰性氣氛下,開始加熱升溫至100℃,反應2小時即可得到親水pa6sip嵌段。採用相同的方法以己二酸(1.2g)代替間苯二甲酸-5-磺酸鈉合成疏水的pa66鏈段。
2)隨後,按pa6sip:pa66質量比1:4迅速的把pa6sip嵌段溶液與pa66嵌段溶液混合轉移到反應球瓶中。同時補加無水氯化鈣補償整個反應體系的損失。在惰性氣氛下,反應在100℃繼續反應24小時。最後得到的共聚醯胺溶液用250ml甲醇沉澱。靜置過夜後,過濾可以得到纖維粉末狀產物,並用甲醇洗滌產物三次。收集到的超聲響應性多嵌段共聚醯胺在90℃下,真空乾燥24小時。為了方便起見,將製備的多嵌段共聚醯胺記為mbcpa。圖1是實施例1製得的mbcpa的核磁圖譜,該圖譜顯示成功合成mbcpa聚醯胺。
3)將3mg多嵌段共聚醯胺溶於6ml的去離子水中,在60℃下,加熱攪拌12小時,最後得到超聲刺激響應性聚醯胺囊泡溶液。圖2是實施例1製得的囊泡的透射電鏡圖。
實施例2
1)在50ml的反應球瓶中加入0.2g的無水硫酸銅,7.0ml的n-甲基吡咯烷酮與1ml的吡啶的混合物溶液,緩慢加熱至60℃,攪拌使之完全溶解。隨後再向溶液中加入2.0g對苯二甲酸-5-磺酸鈉和8.2ml亞磷酸三苯酯,待其完全溶解後,再加入稍過量的1.4g丁二胺。隨後關閉控溫儀,待溶液冷卻後,再抽排三次,用氮氣置換出體系中的空氣。在保持體系惰性氣氛下,開始加熱升溫至95℃,反應10小時即可得到親水pa6sip嵌段。採用相同的方法用己二酸(2.0g)代替對苯二甲酸-5-磺酸鈉合成疏水鏈段。
2)隨後,按親水鏈段:疏水鏈段質量比2:5迅速的把親水和疏水鏈段混合到反應球瓶中。同時補加0.05g無水硫酸銅補償整個反應體系的損失。在惰性氣氛下,反應在120℃繼續反應48小時。最後得到的共聚醯胺溶液用250ml甲醇沉澱。靜置過夜後,過濾可以得到纖維粉末狀產物,並用甲醇洗滌產物三次。收集到的超聲響應性多嵌段共聚醯胺在95℃下,真空乾燥24小時。
3)將6mg多嵌段共聚醯胺溶於6ml的去離子水中,在40℃下,加熱攪拌12小時,最後得到超聲刺激響應性聚醯胺囊泡溶液。
實施例3
1)在50ml的反應球瓶中加入0.4g的無水硫酸鈣,8.0ml的n-甲基吡咯烷酮與3ml的吡啶的混合溶液,緩慢加熱至40℃,攪拌使之完全溶解。隨後再向溶液中加入3.0g間苯二甲酸-5-磺酸鈉(5-ssipa)和6.6ml磷酸三苯酯,待其完全溶解後,再加入稍過量的0.6g己二胺。隨後關閉控溫儀,待溶液冷卻後,再抽排三次,用氮氣置換出體系中的空氣。在保持體系惰性氣氛下,開始加熱升溫至95℃,反應20小時即可得到親水聚醯胺嵌段。用相同的方法,用丁二酸(1.5g)代替間苯二甲酸-5-磺酸鈉(5-ssipa)合成疏水聚醯胺鏈段。
2)隨後,按親水鏈段:疏水鏈段質量比3:5迅速的將親水鏈段和疏水鏈段混合到反應球瓶中。同時補加0.05g無水硫酸鈣補償整個反應體系的損失。在惰性氣氛下,反應在110℃繼續反應72小時。最後得到的共聚醯胺溶液用250ml甲醇沉澱。靜置過夜後,過濾可以得到纖維粉末狀產物,並用甲醇洗滌產物三次。收集到的超聲響應性多嵌段共聚醯胺在105℃下,真空乾燥24小時。
3)將60mg多嵌段共聚醯胺溶於6ml的去離子水中,在50℃下,加熱攪拌12小時,最後得到超聲刺激響應性聚醯胺囊泡溶液。
實施例4
1)在50ml的反應球瓶中加入0.6g的無水氯化鈣,9.0ml的n-甲基吡咯烷酮和4ml的吡啶,緩慢加熱至50℃,攪拌使之完全溶解。隨後再向溶液中加入2.42g對苯二甲酸-5-磺酸鈉和6.5ml磷酸三甲酯,待其完全溶解後,再加入稍過量的1.8g葵二胺。隨後關閉控溫儀,待溶液冷卻後,再抽排三次,用氮氣置換出體系中的空氣。在保持體系惰性氣氛下,開始加熱升溫至105℃,反應20小時即可得到親水鏈段段。用相同的方法,以壬二酸(1.3g)代替對苯二甲酸-5-磺酸鈉合成疏水聚醯胺鏈段。
2)隨後,按親水鏈段:疏水鏈段質量比1:4迅速的將親水鏈段和疏水鏈段混合到反應球瓶中。同時補加0.05g無水氯化鈣補償整個反應體系的損失。在惰性氣氛下,反應在130℃繼續反應96小時。最後得到的共聚醯胺溶液用250ml甲醇沉澱。靜置過夜後,過濾可以得到纖維粉末狀產物,並用甲醇洗滌產物三次。收集到的超聲響應性多嵌段共聚醯胺在100℃下,真空乾燥24小時。
3)將12mg多嵌段共聚醯胺溶於6ml的去離子水中,在60℃下,加熱攪拌12小時,最後得到超聲刺激響應性聚醯胺囊泡溶液。
二、實施例1-4所述的囊泡的藥物釋放能力測試
選用不溶於水的尼羅紅分子作為模型藥物分子測試所製備囊泡的超聲響應特性,對囊泡水溶液而言,如果包裹在囊泡疏水壁中的尼羅紅分子被釋放到水溶液中,由於尼羅紅分子不溶於水,會聚集沉澱下來,從而導致囊泡水溶液中尼羅紅染料螢光強度的降低,因此可以通過檢測囊泡水溶液的螢光光譜,來確定mbcpa囊泡的藥物釋放能力。
圖3是實施例1中的在不同超聲作用時間後囊泡溶液中尼羅紅紫外吸收強度的測試結果。圖4是超聲作用後囊泡溶液圖。由圖3及圖4可以明顯看到囊泡在超聲的刺激下會發生破裂。
表1是實施例1-4囊泡溶液在超聲作用下尼羅紅紫外吸收強度下降50%所用時間。
表1
圖5是在超聲作用下,包裹有尼羅紅染料分子的實施例1製得的囊泡及hbpo-star-peo囊泡水溶液的螢光發射光譜;根據包裹有尼羅紅染料的囊泡的螢光發射光譜得到的歸一化曲線。從圖中看出普通的hbpo-star-peo囊泡在超聲作用下不釋放尼羅紅,即不具有超聲響應性,而本發明中製得的囊泡具有超聲響應獨特性。
圖6是實施例1製得的囊泡在室溫下、50℃、50℃超聲作用下囊泡的螢光發射光譜歸一化曲線。由圖6可發現本發明所述囊泡在遠高於體溫的50℃溫度下也不破裂,說明本發明所述囊泡性能穩定。
以上所述僅是本發明的優選實施方式,應當指出,對於本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護範圍。