面癱復正散的質量檢測方法與流程
2023-05-28 13:18:56 3
本發明涉及中醫藥技術領域,具體涉及面癱復正散的質量檢測方法。
背景技術:
面癱是面神經的非化膿性炎症所致的一種急性周圍性面癱。主要表現為患者面部表情肌癱瘓,即口角歪斜,屬中醫口僻(中經絡證)。《內經》中稱口喁、口僻,並認為口眼歪斜主要是足陽明胃經的病變。明《景嶽全書·痱風》指出:「口眼喁斜有寒熱之辨……然而血氣無虧,則雖熱未必筋緩,雖寒未必筋急,亦總由血氣之衰可知也。」目前,中醫界多認為本病由正氣不足,腠理不密,衛外不固,絡脈空虛,風邪乘虛侵襲面部經絡,面部足陽明經筋失於濡養,氣血痺阻,以致肌肉縱緩不收所致。但有寒熱之別,寒症多有面部受涼因素,熱型面癱多因素體陽盛、寒邪入裡化熱、熱阻陽明、少陽脈絡,致經氣阻滯,經筋失於濡養而發。面癱早期的治療十分關鍵,早期治療是否恰當,直接影響日後的恢復。目前早期治療多給予激素以減輕面神經水腫受壓,減緩面神經變性。早期風熱型患者多伴有感染,所以抗病毒、抗菌素的使用也是十分必要的。中藥外治法治療特發性面神經麻痺療效顯著,大多數研究者配合針灸。
周圍性面癱急性期在臨床最為常見,同時急性期也是治療周圍性面癱的黃金時段,注重此期的治療與調護,可起到事倍功半的療效。採用中藥外用複方製劑,臨床上合併針刺治療,是治療周圍性面癱的一個可行方案。這種治療方法能使藥物直接滲透皮膚,藥效直達病灶,能促進側支循環建立,減輕局部炎性滲出和水腫,改善神經周圍微循環,具有促進局部血流,改善神經營養作用。具有見效快、療程短、效果好、無副反應的優勢。
面癱復正散處方由三七、熟大黃、紫花地丁、厚樸、冰片5味中藥配伍組成,是貴陽中醫學院第二附屬醫院蔡偉副主任醫生根據中醫理論及多年臨床經驗凝集而成用於治療周圍性面癱急性期及局部腫痛患者的純中藥複方外用製劑。其組成藥物分別具有抗炎消腫、抑制炎性滲出、抗菌、鎮痛、降低毛細血管通透性、促進組織創面修復等各方面的功效。臨床上合併針刺治療也表明,面癱復正散對改善耳後疼痛有效,其可能機理是炎症與疼痛呈正相關性,是通過改善患者局部疼痛,使面神經炎的非特異炎性症狀得到有效控制。但目前沒有該藥物適用的質量檢測方法,難以保證製劑的穩定性和療效。
技術實現要素:
本發明的目的是提供面癱復正散的質量檢測方法,以克服現有技術的不足。
本發明的技術方案是這樣的:
面癱復正散的質量檢測方法,該面癱復正散是這樣製備的:將三七5~15份、熟大黃5~15份、紫花地丁10~20份、厚樸5~15份、冰片0.3~1份配齊,除冰片外,三七、熟大黃、紫花地丁和厚樸這四味藥材經60℃減壓乾燥3小時,粉碎成粉末;將冰片研細,與上述粉末配研,過篩,混合,分裝,即得;
該面癱復正散的質量檢測方法包括如下幾項:
(1)性狀:土黃色粉末;氣辛,味苦;
(2)鑑別:包括三七、熟大黃、紫花地丁、厚樸的薄層色譜鑑別;
(3)檢查:包括粒度、外觀均勻度、水分、裝量差異和微生物限度的檢查;
(4)含量測定:包括人參皂苷Rg1的含量測定。
其中,三七的薄層色譜鑑別方法包括如下步驟:
分別製備供試品溶液、陰性對照溶液、對照藥材溶液和對照品溶液;照薄層色譜法試驗,吸取供試品溶液、陰性對照溶液、對照藥材溶液和對照品溶液點於同一矽膠G薄層板上,以三氯甲烷-乙酸乙酯-甲醇-水(15:40:22:10)10℃以下放置的下層溶液為展開劑,展開,取出,晾乾,噴以硫酸乙醇溶液,加熱至斑點顯色清晰;
供試品色譜中,在與對照藥材和對照品色譜相應的位置上,顯相同顏色的螢光斑點,在缺三七藥材的陰性樣品色譜相應位置上,無此斑點。
進一步的,熟大黃的薄層色譜鑑別方法包括如下步驟:
分別製備為供試品溶液、陰性對照溶液和對照藥材溶液;照薄層色譜法試驗,分別取對照藥材溶液、陰性對照溶液和供試品溶液,點於同一矽膠G薄層板上,以正己烷-乙酸乙酯-甲酸(27:10:2.4)為展開劑展開,取出,晾乾,於365nm紫外光燈下檢視;
供試品色譜中,在與對照藥材同位置有相同顏色斑點,陰性對照無幹擾。
進一步的,紫花地丁的薄層色譜鑑別方法包括如下步驟:
分別製備供試品溶液、陰性對照溶液和對照藥材溶液;照薄層色譜法試驗,吸取供試品溶液、陰性對照溶液和對照藥材溶液,分別點於同一矽膠G薄層板上,以甲苯-乙酸乙酯-甲酸(5:3:1)的上層溶液為展開劑,展開,取出,晾乾,置於365nm紫外光燈下檢視;
供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色的螢光斑點,陰性對照無幹擾;
進一步的,厚樸的薄層色譜鑑別方法包括如下步驟:
分別製備供試品溶液、陰性對照溶液和對照藥材溶液;照薄層色譜法,分別吸取供試品溶液、陰性對照溶液和對照藥材溶液,分別點於同一矽膠G薄層板上,以石油醚(60~90℃)-乙酸乙酯-甲酸(8.5:1.5:0.2)為展開劑,展開,取出,晾乾,噴以香草醛硫酸溶液,加熱至斑點顯色清晰;
供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色斑點,且陰性對照無幹擾。
更進一步的,採用平皿法來進行面癱復正散的微生物限度檢查。
更進一步的,所述人參皂苷Rg1的含量測定包括如下步驟:
以本品製備供試品溶液,以人參皂苷Rg1製備對照品溶液,吸取對照品溶液與供試品溶液注入液相色譜儀進行測定,色譜條件為:以Aglient ZORBAX SB-C8(150×4.6mm,5μm)為色譜柱;以乙腈為流動相A,以水為流動相B,進行梯度洗脫;流速為1ml/min;柱溫30℃;檢測波長為203nm;梯度洗脫規定如下:
流動相A:0~20分鐘:18%;20~30分鐘:18→19%;
流動相B:0~20分鐘:82%;20~30分鐘:82→81%。
本發明的技術效果如下:
通過以上方法,能夠制定出面癱復正散的質量標準,從而實現對面癱復正散的質量檢測,經實驗證明,以該方法為標準檢測面癱復正散,可以很好的保證製劑的穩定性,從而保證其療效。
附圖說明
圖1是本發明的製備方法的工藝流程圖;
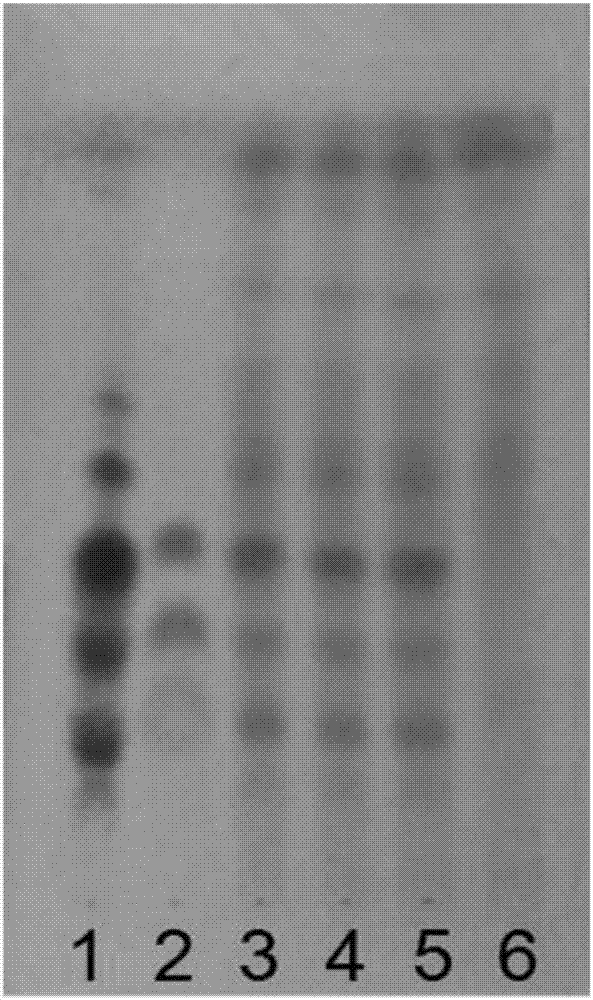
圖2是本發明的三七的薄層鑑別圖;圖中,1、三七對照藥材;2、三七皂苷R1和人參皂苷Rg1和人參皂苷Rb1混合對照品;3~5、面癱復正散供試品(批號:20160311,20160312,20160313);6、陰性供試品(缺三七藥材);
圖3是熟大黃的薄層鑑別圖;圖中:1、熟大黃對照藥材;2~4、面癱復正散供試品(批號:20160311,20160312,20160313);5、陰性供試品(缺熟大黃藥材);
圖4是紫花地丁的薄層鑑別圖;圖中,1、紫花地丁對照藥材;2~4、面癱復正散供試品(批號:20160311,20160312,20160313);5、陰性供試品(缺紫花地丁藥材);
圖5是厚樸的薄層鑑別圖;圖中,1、厚樸對照藥材;2~4、面癱復正散供試品(批號:20160311,20160312,20160313);5、陰性供試品(缺厚樸藥材);
圖6是厚樸的薄層鑑別圖一;圖中,1、冰片對照藥材;2、面癱復正散供試品(批號:20160501);3、陰性供試品(缺冰片藥材);
圖7是厚樸的薄層鑑別圖二;圖中,1、冰片對照藥材;2、面癱復正散供試品(批號:20160501);3、陰性供試品(缺冰片藥材)。
具體實施方式
下面結合附圖和實施例對本發明作進一步的詳細說明,但本發明的保護範圍不受具體實施例的任何限制,而是由權利要求加以限定。
實施例1:
1、處方組成:三七10g 熟大黃10g 紫花地丁15g 厚樸10g冰片0.5g。
2、製備方法:如圖1所示的工藝流程,將以上五味,按處方23倍量配齊,除冰片外,三七、熟大黃等四味藥材經60℃減壓乾燥3小時,粉碎成最細粉;將冰片研細,與上述粉末配研,過篩,混合,分裝,即得。裝量標準:6g/袋。
3、功能主治:清熱消腫、活血通絡。用於周圍性面癱急性期及局部腫痛患者。
4、用法用量:外用,根據病情,選用凡士林、油脂等調勻外敷,帖敷局部穴位,每穴一貼,每日一次,1月為1療程。根據病情需要可再重複療程。
5、檢查:按照散劑項下的有關規定(《中國藥典》2015年版四部通則0115)進行檢查。分別檢查粒度、外觀均勻度、水分、裝量差異、微生物限度,應符合規定。
5.1、粒度:按照散劑「粒度」項下(通則0115)測定,應符合規定。
5.2、外觀均勻度按照散劑「外觀均勻度」項下(通則0115)測定,應符合規定。
5.3、水分:按照水分測定法項下第二法(《中國藥典》2015年版四部通則0832)進行測定,水分不得過9.0%。
5.4、裝量差異:取本品10袋,照散劑「裝量差異」項下(通則0115)依法檢查,裝量差異限度應在±7%以內,超出裝量差異限度的不得多於2袋,並不得有1袋超出限度1倍。
實驗例1:
面癱復正散的質量檢測方法:
1性狀:中試三批樣品均為土黃色粉末;氣辛,味苦。
2薄層鑑別:
2.1三七的薄層色譜研究
取本品2g,加5ml水,攪勻,再加水飽和的正丁醇50ml,超聲處理1小時,濾過,濾液用120ml正丁醇飽和的水萃取,分取正丁醇層,水浴蒸乾,殘渣用1ml甲醇溶解,作為供試品溶液。取三七的陰性模擬製劑2g,同法製備陰性對照溶液。另取三七藥材粉末0.5g,同法製成對照藥材溶液。再稱取人參皂苷Rg1對照品、人參皂苷Rb1對照品及三七皂苷R1對照品適量,分別用甲醇製成每1ml 含0.5mg的溶液,作為對照品溶液。照薄層色譜法(《中國藥典》2015年版四部通則0502)試驗,吸取上述四種溶液各5μl,分別點於同一矽膠G薄層板上,以三氯甲烷-乙酸乙酯-甲醇-水(15:40:22:10)10℃以下放置的下層溶液為展開劑,展開,取出,晾乾,噴以10%的硫酸乙醇溶液,在105℃加熱至斑點顯色清晰。供試品色譜中,在與對照藥材和對照品色譜相應的位置上,顯相同顏色的斑點,在缺三七藥材的陰性樣品色譜相應位置上,無此斑點(見圖2)。
2.2熟大黃的薄層色譜研究
取本品2g,加甲醇15ml,超聲提取20min,濾過,濾液濃縮至1ml,作為供試品溶液。取缺熟大黃的陰性模擬製劑2g,同法製備陰性對照溶液。另取熟大黃對照藥材粉末5g,加40ml水直火回流提取1h,濾液水浴上揮幹,殘渣加甲醇10ml轉入錐形瓶中,超聲提取10min,濾液濃縮至1ml,作為對照藥材溶液。照薄層色譜法(《中國藥典》2015年版四部通則0502)試驗,分別取對照藥材溶液3μl、陰性對照溶液、複方供試溶液各2μl,點於同一矽膠G薄層板上,以正己烷-乙酸乙酯-甲酸(27:10:2.4)為展開劑展開,取出,晾乾,於365nm紫外光燈下檢視。供試品色譜中,在與對照藥材同位置有相同顏色斑點,陰性對照無幹擾(見圖3)。
2.3紫花地丁的薄層色譜研究
取本品2g,加甲醇20ml溶解,超聲處理30min,濾過,濾液蒸乾,殘渣加純水10ml,攪拌使溶解,濾過,濾液蒸乾,殘渣加乙酸乙酯1ml使溶解,作為供試品溶液。取缺紫花地丁的陰性模擬製劑2g,同法製備陰性對照溶液。另取紫花地丁對照藥材1.0g,同法製成對照藥材溶液。照薄層色譜法(《中國藥典》2015年版四部通則0502)試驗,吸取上述三種溶液各3μl,分別點於同一矽膠G薄層板上,以甲苯-乙酸乙酯-甲酸(5:3:1)的上層溶液為展開劑,展開,取出,晾乾,置於紫外燈(365nm)下檢視。供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色的螢光斑點,陰性對照無幹擾(見圖4)。
2.4厚樸的薄層色譜研究
取供試品2g,取按配方比例配製的陰性對照樣品2g,分別加甲醇15ml,超聲30min,濾過,濾液分別作為供試品溶液和陰性對照溶液。另取厚樸對照藥材粉末0.5g,加5ml甲醇,同法製成對照藥材溶液;照薄層色譜法(《中國藥典》2015年版四部通則0502)試驗,分別吸取上述供試品溶液、陰性對照溶液各2μl,對照藥材溶液5μl,分別點於同一矽膠G薄層板上,以石油醚(60~90℃)-乙酸乙酯-甲酸(8.5:1.5:0.2)為展開劑,展開,取出,晾乾,噴以1%香草醛硫酸溶液,在100℃加熱至斑點顯色清晰。供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色斑點,且陰性對照無幹擾(見圖5)。
2.5冰片的薄層色譜研究
參照「TLC、GC法檢測冠心蘇合膠囊中的冰片」[14]中對冰片薄層色譜研究:取本品1g,加入乙酸乙酯10ml,萃取,取乙酸乙酯液,作為供試品溶液。取缺冰片陰性模擬試劑1g,同法製備陰性對照液。另取冰片對照藥材1g,加乙酸乙酯製成濃度為2mg/ml的溶液,作為對照品溶液。照薄層色譜法(《中國藥典》2015年版四部通則0502)試驗,吸取上述三種溶液各2μl,分別點於矽膠G薄膜板上,以石油醚(30~60℃)-乙酸乙酯(9:1)為展開劑,展開,取出,晾乾,噴以磷鉬酸試液,熱風吹至斑點顯色清晰。供試品色譜中,在與對照藥材色譜相應的位置上,無相應的斑點(見圖6)。參照「複方消炎散中冰片和薑黃的薄層色譜鑑別」中對冰片薄層色譜研究:取本品1g,加入三氯甲烷10ml萃取,取三氯甲烷液,作為供試品溶液。取缺冰片陰性模擬試劑1g,同法製備陰性對照液。另取冰片對照藥材,加乙酸乙酯製成濃度為1mg/ml的溶液,作為對照品溶液。照薄層色譜法(《中國藥典》2015年版四部通則0502)試驗,吸取上述三種溶液各2μl,分別點於矽膠G薄層板上,以環己烷-乙酸乙酯(17:3)為展開劑,展開,取出,晾乾,噴以磷鉬酸試液,熱風吹至斑點顯色清晰。供試品色譜中在與對照藥材色譜相應的位置上,無相應的斑點(見圖7)。故冰片的定性鑑別未列入本發明的質量檢測方法。
實驗例2:微生物限度檢查法
1、供試液的製備:取本品10ml,加入不超過45℃PH7.0無菌氯化鈉-蛋白腖稀釋液至100ml,邊加邊攪拌,使供試品充分混勻,即得1:10的供試液。
2、微生物計數法:需氧菌總數計數:取1:10供試液1ml分注1個皿,1ml/皿,按《中國藥典》2015版四部非無菌產品微生物限度檢查:微生物計數法(通則1105)項下平皿法進行菌落計數。
黴菌和酵母菌總數計數:取1:10供試液2ml分注2個皿,1ml/皿,按《中國藥典》2015版四部非無菌產品微生物限度檢查:微生物計數法(通則1105)中的平皿法進行菌落計數。
3、控制菌檢查:
金黃色葡萄球菌:取1:10供試液按照《中國藥典》2015版四部非無菌產品微生物限度檢查:控制菌檢查法(通則1106)下相應控制菌檢查方法進行。
銅綠假單胞菌:取1:10供試液按照《中國藥典》2015版四部非無菌產品微生物限度檢查:控制菌檢查法(通則1106)下相應控制菌檢查方法進行。
藥品微生物限度檢查方法適用性試驗報告
培養基、稀釋劑及來源:
0.9%無菌氯化鈉-蛋白腖緩衝液、胰酪大豆腖瓊脂、沙氏葡萄糖瓊脂、胰酪大豆腖液體培養基、麥康凱液體培養基、麥康凱瓊脂培養基等。(上海盛思生化科技有限公司生產,均按《中國藥典》2015版相關規定配製及滅菌)。
菌種及來源:
以上菌種均為中國食品藥品檢定研究院派生菌種。
實驗儀器:高壓蒸汽滅菌器、電子天平(十分之一)、潔淨室淨化工作檯,菌落計數器等。
結果:適用性記錄及結果如下:
1、需氧菌、黴菌及酵母菌總數計數方法選擇試驗結果見表1;
2、需氧菌、黴菌及酵母菌總數計數方法驗證回收試驗見表2-4(附表記錄1-3);
3、控制菌——金黃色葡萄球菌檢查方法的驗證結果見下表5;
4、控制菌——銅綠假單胞菌檢查方法的驗證結果見下表6;
表1需氧菌、黴菌及酵母菌總數計數方法選擇試驗結果
備註:「——」表示未做此項。
結論:
1、1:10的供試液採用平皿法(1ml/皿)大腸埃希菌、白色念珠菌、黑麴黴的回收率能達到70%以上;1:10的供試液採用平皿法(0.5ml/皿)枯草芽孢桿菌的回收率能達到70%以上,1:10的供試液採用平皿法(0.2ml/皿)金黃色葡萄球菌的回收率能達到70%以上。
2、面癱復正散具有抑菌活性成分(如:紫花地丁、厚樸等),因此在進行微生物驗證時,出現較明顯的抑菌能力,尤其是對金黃色葡萄球菌的抑制效果明顯,其次是對枯草芽孢桿菌有一定的抑制作用,所以採用平皿法進行,而藥物對枯草芽孢桿菌的作用不是很明顯,採取常規法即可。根據驗證結果,綜合考慮驗證操作步驟,最終確定採用平皿法(1:10供試液1ml,分注5個平皿)來進行面癱復正散的微生物限度檢查。
表2需氧菌、黴菌及酵母菌總數計數方法驗證回收試驗記錄1
註:試驗組:供試液+試驗菌液;供試品對照組:供試液+稀釋液;菌液組:稀釋液+試驗菌液。
表3需氧菌、黴菌及酵母菌總數計數方法驗證回收試驗記錄2
表4需氧菌、黴菌及酵母菌總數計數方法驗證回收試驗記錄3
表5金黃色葡萄球菌檢查方法的驗證試驗記錄
備註:甘露醇氯化鈉平板中:「+」標示有典型菌落生長;「—」標示無菌生長。
血漿凝固酶試驗中:「+」標示有血漿凝固;「—」標示血漿不凝固。
「——」表示未做此項。
表6銅綠假單胞菌檢查方法的驗證試驗記錄
備註:甘露醇氯化鈉平板中:「+」標示有典型菌落生長;「—」標示無菌生長。
氧化酶試驗中:「+」標示紙上培養物出現粉紅色—紫紅色的變化;「—」標示紙上培養物不變色或僅顯粉紅色改變。
綠膿菌素試驗中:「+」標示出現粉紅色變色反應;「—」標示無粉紅色出現。
「——」表示未做此項。
結論:
經本試驗驗證,面癱復正散的需氧菌計數可按《中國藥典》2015版四部通則1105項下平皿法(1:10供試液,0.2ml/皿)進行計數;黴菌及酵母菌總數計數可按《中國藥典》2015版通則1105項下平皿法(取1:10供試液2ml分注2個皿,1.0ml/皿)計數;控制菌中金黃色葡萄球菌和銅綠假單胞菌的檢查可按《中國藥典》2015版通則1106項下規定的常規法進行檢查。
實驗例3:含量測定
人參皂苷Rg1照高效液相色譜法(《中國藥典》2015年版四部通則0512)測定。
1.儀器與試藥
儀器:Agilent 1260高效液相色譜儀;梅特勒-託利多AE電子分析天平(METTLER TOLEDO十萬分之一)。
試藥:乙腈[色譜純,美國天地(TEDIA)有限公司];水(娃哈哈飲用純淨水,貴陽娃哈哈食品有限公司製造)。
對照品:人參皂苷Rg1對照品(供含量測定用,含量以91.7%計,批號為:110703-201530),購自中國食品藥品檢定研究所。
樣品:面癱復正散(批號:20160501 20160502 20160503)。
2.對照品溶液製備
對照品溶液的製備:精密稱取人參皂苷Rg1對照品4.536mg,加甲醇製成每1ml含人參皂苷Rg1 0.416mg的溶液,即得。
3.色譜條件的選擇:
在進行人參皂苷Rg1色譜條件選擇時,參考文獻,進行了色譜柱、流動相以及柱溫的篩選。
3.1色譜柱的選擇
不同的色譜柱對化學成分分離有一定的影響,在此考察了Diamonsil Plus C18色譜柱(250×4.6mm,5μm);Ultimate XB-C18色譜柱(250×4.6mm,5μm);Aglient ZORBAX SB-C18(250×4.6mm,5μm)及Aglient ZORBAX SB-C8(150×4.6mm,5μm)色譜柱四種,結果見表7。
表7人參皂苷Rg1色譜柱條件選擇表
通過對不同色譜柱條件下色譜圖進行比較,結果表明Ultimate XB-C18色譜柱峰前延,對稱性差,分離度低;Diamonsil plus C18色譜柱峰展寬,峰形差;Agilent Zorbax SB-C18色譜柱峰拖尾,分離效果差;Agilent Zorbax SB-C8色譜柱峰形較好,分離度較高,因此確定Agilent Zorbax SB-C8(150×4.6mm,5μm)色譜柱為試驗用柱。
3.2流動相的考察
對三種不同洗脫系統進行考察,結果見表8-10。
表8洗脫系統一
表9洗脫系統二
表10洗脫系統三
通過比較以上三種不同洗脫系統的色譜峰,其中洗脫系統一人參皂苷Rg1與其他組分峰未完全分離,分離效果差;洗脫系統二色譜峰分離較好,峰形較銳;洗脫系統三人參皂苷Rg1與其他組分峰未完全分離,且陰性有幹擾,分離效果差。因此確定洗脫系統二為流動相條件。
3.3柱溫的考察
分別考察了25℃、30℃、35℃時色譜分離效果。
結果表明,柱溫為25℃時,人參皂苷Rg1與其他組分峰未完全分離,分離效果差;柱溫為30℃、35℃對色譜峰的分離效果影響不明顯,考慮到室溫等情況,選擇柱溫為30℃。
綜上,確定人參皂苷Rg1的色譜條件為Agilent Zorbax SB-C8(150×4.6mm,5μm)色譜柱;乙腈-水梯度洗脫:0~20min(18:82),21~30min(19:81);流速:1ml/min;柱溫:30℃;檢測波長:203nm;分析時間:30min;進樣量:10μl。理論板數按人參皂苷Rg1計算應不低於4000。
當然,以上只是發明的具體應用範例,本發明還有其他的實施方式,凡採用等同替換或等效變換形成的技術方案,均落在本發明所要求的保護範圍之內。