一種Au@MnPS催化劑及其製備方法和用途與流程
2023-06-01 03:03:21 3
本發明涉及一種Au@MnPS3催化劑的製備方法及其在環己烯環氧化中的應用。
背景技術:
環己烯選擇性氧化是合成多種高附加值中間體環己烯醇,環己烯酮和環氧環己烷的重要工藝。由於環己烯分子中含有一個易發生氧化反應的不飽和C=C雙鍵和多個活性α-H原子,導致發生氧化反應的選擇性較差,產物複雜,學術界和工業界一直在尋求溫和條件下具有高活性、高選擇性的催化劑用於環己烯催化氧化反應。多相催化劑具有容易與產物分離和便於循環使用的優勢,是綠色催化工藝的主要研究方向之一。
新型的多相催化劑涵蓋諸多方向,對於選擇性氧化來說,當下的研究熱點之一便是納米金催化劑。納米金催化劑現在已經有用於多種選擇性氧化的報導,CO氧化、丙烯環氧化、氫氧直接合成過氧化氫、環己烷氧化制KA油等,其中也包括在環己烯選擇性氧化中的應用。而納米金催化劑的載體一般為多孔材料,包括活性炭,分子篩等等。在多孔材料的方面,近年來,金屬-有機骨架配合物(metal - organic frameworks,MOFs)逐漸走進人們的視野,但現在絕大多數MOF的相關研究工作都集中在該材料對氣體的吸附,催化方面的應用研究也才剛開始嶄露頭角。MOFs作為載體具有比表面積大,可方便地調控其結構性質(如孔徑的擴張或壓縮)及表面功能基團,具有不飽和金屬配位點等優點。由於MOFs能夠有效的防止反應過程中顆粒的長大,有許多MOFs負載金屬納米催化劑在有機反應中表現出了優秀的催化性能,並且也有了Au@MOFs的報導。然而現在的Au@MOFs的研究基本上只集中在醇類氧化上,基本沒有用於烯烴氧化的研究。
由於仿生催化劑一直在環己烯的選擇性氧化中表現不俗,而金屬卟啉可以形成構成MOFs的金屬有機配體結構,因此MOFs 材料是實現仿生催化劑均相催化劑多相化手段之一。MOFs 的特殊結構使得仿生催化劑中有機配體的部分通過金屬離子連接以單分子態存在,彼此互相分離,避免了二聚,可提高穩定性和催化活性。同時,其均勻的孔徑和規整的孔道結構,又為催化反應提供良好的形狀選擇性。PIZA-3是由Suslick等最早報導的卟啉作為有機配體製備的MOFs 材料,有能力在亞碘醯苯為氧化劑的條件下催化烯烴環氧化,過氧化物產率可達20-74%。可以看出,金屬卟啉MOFs在環己烯的選擇性氧化方面具有巨大的潛力。
技術實現要素:
為了克服現有技術的不足,本發明目的在於提供一種Au@MnPS催化劑及其製備方法和用途。
我們嘗試將納米金和金屬卟啉MOFs相結合,合成了錳卟啉的金屬有機框架MnPS,並將其作為載體製備了Au@MnPS催化劑用於環己烯氧化反應,對催化劑進行表徵並考察不同反應條件對催化劑性能的影響。在文獻中並沒有將金屬卟啉MOFs用作載體的先例,單獨使用納米金作為活性中心的催化劑在環己烯的氧化中並不能很好的獲得環氧環己烷,另外金屬卟啉MOFs用於催化氧化時所用的氧化劑多為亞碘醯苯和TBHP。本發明將納米金引入金屬卟啉MOFs後,納米金可以活化分子氧提高反應的轉化率,而金屬卟啉MOFs則可以與此同時保證反應的選擇性,兩者的結合起到了比分別使用更好的技術效果。
一種Au@MnPS催化劑,它由Au和MnPS組成,其中Au和MnPS中的錳卟啉結構作為催化劑的主要活性成分,Au的質量百分含量為2.0%,MnPS用作催化劑的載體,其質量百分含量>95%。
所述的Au為Au的金屬、Au的金屬氧化物或上述兩者的混合物。
所述的MnPS為一種以錳卟啉結構為主的金屬有機框架。
一種根據所述的Au@MnPS催化劑的製備方法,包括如下步驟:
1)將卟啉和十二水合醋酸錳溶於DMF之中,置於水熱合成釜中,在180℃反應48h後,冷卻至室溫,抽濾,用去離子水洗滌,得到黑色塊狀的MnPS固體,研磨至粉末狀;
2)上述步驟1)得到的黑色固體取0.5g,在三口燒瓶中加入尿素0.9g,氯金酸溶液2.5mL,用去離子水稀釋至30mL,在80℃攪拌回流6h後,加入30mL甲醛溶液,50℃繼續反應3h,待反應液冷卻後,抽濾,用去離子水洗滌,110℃乾燥2 h,得到塊狀固體,研磨得到Au@MnPS粉末。
一種根據所述的Au@MnPS催化劑的用途,用於環己烯直接選擇性環氧化生成環氧環己烷。
本發明與現有技術相比具有的有益效果:
1)環己烯環氧化的轉化率和選擇性較好;
2)條件溫和;
3)可以用綠色的分子氧為氧化劑。
附圖說明
圖1是Au@MnPS的TEM圖;
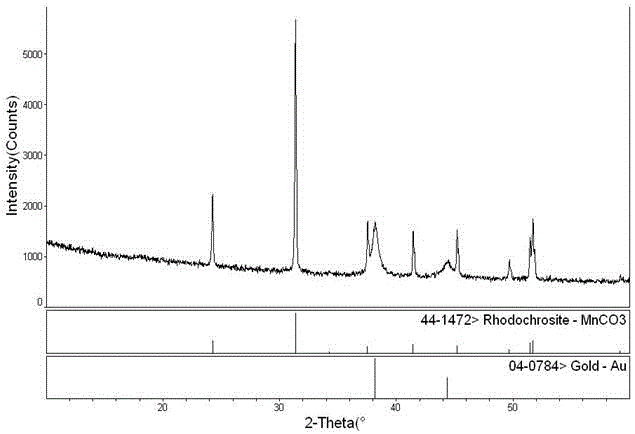
圖2是Au@MnPS的XRD譜圖;
圖3是Au@MnPS的SEM圖;
圖4 是Au@MnPS的EDS譜圖。
具體實施方式
以下結合附圖和實施例對本發明做進一步的說明。
實施例1
Au@MnPS催化劑的製備:
3.8625g香草醛溶於50 mL丙酸中,141℃冷凝回流,緩慢滴加1.3 mL吡咯後,反應45min。冷卻至室溫,抽濾,並用無水甲醇洗滌數次。得到的紫紅色固體在110℃乾燥2h;將製得的卟啉0.2415g和醋酸錳1.2255g溶於DMF之中,放入水熱合成釜中。在180℃反應48h後,冷卻至室溫,抽濾,用去離子水洗滌,得到黑色塊狀的MnPS固體,研磨至粉末狀。取上述步驟得到的黑色固體0.5g,在三口燒瓶中加入尿素0.9g,根據金負載量不同加入不同量的氯金酸溶液(HAuCl4·4H2O,濃度為0.1g/L),用去離子水稀釋至30mL。在80℃攪拌回流6h後,加入30mL市售甲醛溶液(國藥集團化學試劑有限公司),50℃繼續反應3h。待反應液冷卻後,抽濾,用去離子水洗滌,110℃乾燥2 h,得到塊狀固體,研磨得到不同金含量Au@MnPS粉末。其中得到的金含量2%Au@MnPS的催化劑XRD如圖2所示,TEM圖如圖1所示,SEM圖如圖3所示,EDS圖如圖4所示。
催化劑活性評價中產物採用氣相色譜分析,毛細管柱為SE-54 (30 m×0.32mm×0.5μm),檢測器為火焰離子化檢測器(FID)。正己烷作為內標物。
以分子氧為氧化劑,採用DP法製備的錳分子篩負載納米金催化劑對於環己烷選擇性氧化,在80 ℃、0.5 MPa、24 h、無溶劑反應條件下,環己烯氧化轉化率為39.9%,環氧環己烷選擇性僅為2.52%;採用CZJ-1-Mn(一種錳卟啉MOFs)作為催化劑時,在乙腈溶劑,室溫反應6h條件下,雖然環己烯轉化率和環氧環己烷選擇性都有99%,但是所用的氧化劑為亞碘醯苯,和分子氧相比不夠綠色。
實施例2
催化劑活性評價。金含量2%Au@MnPS催化劑0.10 g置於一個PTFE-lined高壓釜(容積= 20mL)中,0.8215g的環己烯,溶劑乙腈10mL,TBHP引發劑5滴,和作為氧化劑的分子氧在1 MPa的壓力下在80°C攪拌反應20小時。得到環己烯氧化的轉化率為45.047%,環氧環己烷的選擇性為68.406%,環己烯醇的選擇性為12.502%,環己烯酮的選擇性為12.675%,鄰環己二醇的選擇性為6.416%。
實施例3
催化劑活性評價。金含量2% Au@MnPS催化劑0.10 g用於反應。0.8215g的環己烯,溶劑乙腈10mL,TBHP引發劑5滴,和作為氧化劑的分子氧在1 MPa的壓力下在80°C攪拌反應5小時。得到環己烯氧化的轉化率為39.667%,環氧環己烷的選擇性為52.966%,環己烯醇的選擇性為9.566%,環己烯酮的選擇性為10.827%,鄰環己二醇的選擇性為26.641%。
實施例4
催化劑活性評價。金含量2% Au@MnPS催化劑0.10 g用於反應。0.8215g的環己烯,溶劑乙腈10mL,TBHP引發劑5滴,和作為氧化劑的分子氧在1 MPa的壓力下在80°C反應攪拌15小時。得到環己烯氧化的轉化率為71.942%,環氧環己烷的選擇性為76.585%,環己烯醇的選擇性為7.013%,環己烯酮的選擇性為11.302%,鄰環己二醇的選擇性為5.101%。
實施例5
催化劑活性評價。金含量2% Au@MnPS催化劑0.10 g用於反應。0.8215g的環己烯,溶劑乙腈10mL,TBHP引發劑5滴,和作為氧化劑的分子氧在1 MPa的壓力下在120°C攪拌反應20小時。得到環己烯氧化的轉化率為77.904%,環氧環己烷的選擇性為71.722%,環己烯醇的選擇性為2.439%,環己烯酮的選擇性為6.628%,鄰環己二醇的選擇性為19.211%。
實施例6
催化劑活性評價。金含量0.25% Au@MnPS催化劑0.10 g用於反應。0.8215g的環己烯,溶劑乙腈10mL,TBHP引發劑5滴,和作為氧化劑的分子氧在1 MPa的壓力下在80°C攪拌反應20小時。得到環己烯氧化的轉化率為88.439%,環氧環己烷的選擇性為82.967%,環己烯醇的選擇性為1.924%,環己烯酮的選擇性為1.209%,鄰環己二醇的選擇性為13.900%。
實施例7
催化劑活性評價。金含量2% Au@MnPS催化劑0.10 g用於反應。0.8215g的環己烯,溶劑乙腈10mL,TBHP引發劑5滴,和作為氧化劑的分子氧在1 MPa的壓力下在80°C無攪拌反應20小時。得到環己烯氧化的轉化率為37.095%,環氧環己烷的選擇性為75.617%,環己烯醇的選擇性為3.921%,環己烯酮的選擇性為6.017%,鄰環己二醇的選擇性為14.444%。