一種離子液體輔助微波輻射法合成超小磁性納米簇的方法與流程
2023-06-04 11:16:07 1
本發明涉及納米材料的製備技術領域,尤其是涉及一種離子液體輔助微波輻射法合成超小磁性納米簇的方法。
背景技術:
我國是發現磁現象最早的國家,而且很早就將磁性物質引入疾病的治療領域,在傳統中醫中就有磁石可「潛陽納氣,鎮驚安神」的理論。隨著近現代醫學技術和磁性理論的發展,磁性材料在醫學中的應用也進入了一個新的發展時期。尤其是當前人們對納米材料在生物醫學中應用的研究不斷加深,在靶向給藥、成像檢測、腫瘤熱療等領域納米磁性材料都有著廣泛的應用前景。
製備磁性納米材料目前有多種方法,包括物理製備方法,如高能機械球磨法、物理氣相沉積法;化學製備方法,如微乳液法、高溫分解法等。但製得的磁性納米粒子,在尺寸、均一性、水溶性等方面仍然很難做到盡如人意。為了改善磁性納米粒子的水溶性,並充分發揮其在生物醫學領域的應用潛力,一般需要在磁性納米粒子表面引入羧基(-COOH)、氨基(-NH2)、巰基(-SH)等功能基團。攜帶不同功能基團磁性納米粒子通過與不同的生物大分子等生物活性物質結合,便可展現出各異的功能特性。例如,抗體蛋白質分子中含有大量的氨基,在一定的條件下氨基和羧基可以發生偶聯反應。因此表面攜帶羧基的磁性納米粒子便可通過羧基氨基的偶聯反應與抗體結合在一起,構成免疫磁珠,應用在免疫層析檢測中可大幅度提高檢測技術的靈敏性。
抗壞血酸又稱維生素C,是人體等其他許多動物必不可少的營養素,在生物體內主要起到一種抗氧化的作用。將抗壞血酸引入反應體系,在微波輔助加熱下,抗壞血酸起到抗氧化的作用,保護部分Fe2+離子不被氧化為Fe3+離子,有助於四氧化三鐵磁性納米粒子的形成;同時還通過化學鍵合使得磁性納米粒子表面連接上羧基,增強了磁性納米粒子的水溶性和功能性。
目前,大尺度地製備超小磁性粒子仍然面臨一些困難,如:合成步驟複雜、合成成本高、需要合成油相轉水相的兩親性聚合物、生物相容性差等。
中國專利ZL200810041462.2公開了一種微波製備粒徑可調納米四氧化三鐵微球的方法,其步驟為:第一步,將鐵鹽溶於多元醇中,再加入無機鹽和表面活性劑,以及加入助溶劑,混合後攪拌,得到製備四氧化三鐵的前體液體,其中:多元醇與鐵鹽的質量比5~100∶1,無機鹽與鐵鹽的質量比1~6∶1,表面活性劑與鐵鹽的質量比0~5∶1,助溶劑與多元醇的體積比0~1∶1;第二步,將四氧化三鐵的前體液體置於微波用的玻璃管中,在微波反應器中進行微波加熱反應,反應後得到黑色的磁性四氧化三鐵納米粒子。
綜合已報導的方法,得知極難高效合成具有T1造影效應的超小四氧化三鐵納米粒子的核心科學問題是反應過程中成核和生長速率過快,不能按照晶體結構習性抑制材料的生長,且不能一步法實現四氧化三鐵納米粒子功能化。
技術實現要素:
本發明的目的就是為了克服上述現有技術存在的缺陷而提供一種離子液體輔助微波輻射法合成超小磁性納米簇的方法,以超純水為溶劑,採用生物-離子液體輔助微波輻射法高效合成出超小四氧化三鐵納米簇。本發明專利實質性特點和創新性是:微波輔助合成工藝具有節能高效、工藝簡單、合成尺度大等優點,可顯著提升納米材料的性能,廣泛應用於生物醫學、催化材料、光電子學等領域。同時,生物-離子液體的加入可抑制四氧化三鐵晶體的長大,並使得納米粒子表面功能化。通過該技術路線合成的超小磁性納米簇具有優良的T1造影效果,具有臨床應用前景,為探索材料微觀形貌與應用性質之間的內在關係奠定了技術基礎。
本發明的目的可以通過以下技術方案來實現:
一種離子液體輔助微波輻射法合成超小磁性納米簇的方法,包括以下步驟:
(1)稱取有機鐵鹽和無機亞鐵鹽,分別溶於水,配成溶液;
(2)將步驟(1)製得的兩份溶液混合,再加入抗壞血酸,攪拌,得到混合溶液;
(3)繼續往步驟(2)得到的混合溶液中加入生物離子液體,得到前驅體溶液,所述的生物離子液體為膽汁素-羧酸鹽陰離子液體或PEG化咪唑二烴基磷酸鹽離子液體;
(4)將前驅體溶液轉移到微波反應器中,反應;
(5)將步驟(4)所得反應產物轉移,分離,即得到所述超小磁性納米簇。
步驟(1)中所述的有機鐵鹽為檸檬酸鐵、乙醯丙酮鐵或右旋糖酐鐵中的至少一種;
所述的無機亞鐵鹽為硫酸亞鐵或氯化亞鐵中的至少一種;
加入的有機鐵鹽與無機亞鐵鹽的量滿足Fe2+和Fe3+的摩爾比1:1~4。
步驟(2)中抗壞血酸與Fe2+離子的摩爾比為1:3~20。
所述的膽汁素-羧酸鹽陰離子液體由膽汁素與羧酸鹽按摩爾配比1:1反應,分離純化得到,其具體反應條件為:反應溫度為100-150℃,反應時間為5-20h,分離純化後得到膽汁素-羧酸鹽陰離子液體,收率為80%-95%。其中,羧酸鹽選自檸檬酸鹽、乙酸鹽、琥珀酸鹽或馬來酸鹽中的一種。
所述的PEG化咪唑二烴基磷酸鹽離子液體由PEG與咪唑二烴基磷酸鹽按摩爾配比1:1反應,分離純化得到,其具體反應條件為:反應溫度為70-150℃,反應時間為1-10h,分離純化後得到PEG化咪唑二烴基磷酸鹽離子液體,收率為85%-98%。其中,PEG的未反應端為羥基、氨基、羧基、巰基或酯基中一種(PEG的兩端可以修飾,因此,未反應端的羥基可以相應被修飾為氨基、羧基、巰基或酯基等)。
生物-離子溶液的加入量為混合溶液的0.1wt%~3wt%。
步驟(3)中混合溶液中加入生物-離子液體後,再調節pH至9~12,才得到所述前驅體溶液。
步驟(4)中反應的工藝條件為:微波功率為500~1200W,微波輻射頻率為2450MHz,反應溫度為100-200℃,反應時間為0.5~4h。
步驟(5)的分離工藝為:先將反應產物轉移至透析袋中,透析1~2天,再在10000-14000rpm的離心轉速下,差速離心分離2-10min。
採用上述製備方法製備的離子液體輔助微波輻射法合成超小磁性納米簇,所述的超小磁性納米簇的平均尺寸為2~5nm。
所述的超小磁性納米簇的r1弛豫率為3.5-9s-1mM-1,r2弛豫率為20-35s-1mM-1,r2/r1為0.175-10。
所述的微波輔助加熱將高強度短脈衝微波輻射聚焦到含「敏化劑」(強吸收微波的物質如鐵磁金屬)的四氧化三鐵納米晶核表面,由於表面金屬與微波的強烈相互作用,微波能轉變為熱,使表面上一些點被選擇性地快速加熱至很高的溫度,導致反應器中的有機鐵鹽與受激發的晶核表面接觸發生反應。通過適當控制微波脈衝時間,可控制納米粒子表面的溫度、反應物的壓力和流速,並可進一步控制化學反應,從而有效控制四氧化三鐵的粒徑。同時,微波加熱還具有非熱效應,微波與四氧化三鐵晶體的低頻彈性晶格振動耦合,產生非熱分布,可以提高了離子遷移率,從而加快了載流子向表面的擴散,導致羥基自由基的增加以及表面電子濃度的增大,抑制了四氧化三鐵納米粒子的生長。所述的生物-離子液體,在微波輻射下能夠更好地提供熱量,並且通過靜電作用迅速與鐵離子和亞鐵離子結合,加快四氧化三鐵晶核形成,由於生物-離子液體存在獨特的空間位阻結構、多負電荷和低界面張力性質,極易在晶核表面形成了一個穩定有序的保護層,屏蔽鐵離子和亞鐵離子與晶核接觸,阻礙鐵離子和亞鐵離子與晶核的作用,進而抑制各晶面生長,來控制納米四氧化三鐵粒子尺寸和提高納米粒子的穩定性。得到的2-5nm的超小磁性納米簇具有優良的T1造影效果,具有臨床應用前景。
傳統的單分散高品質磁性納米粒子合成尺度大多在毫克級別,如果提高合成尺度,主要問題是磁性納米粒子粒徑、形貌和磁強度得不到有效控制,而本專利提出的合成方法主要通過原子/分子尺度對前驅體進行形貌和分散性控制,從而保證了前驅體分散、組裝、熱分解過程和磁性粒子最終形成過程。克級尺度規模化合成高品質磁性納米粒子需要提高反應體系的固含量,傳統的製備工藝固含量都相對較少(5%,合成尺度顯著提高,並且磁性納米粒子的粒徑、形貌、分散性和磁強度都能得到有效控制。
本發明中,採用生物-離子液體,可以起到重要結構生長抑制劑及無機納米顆粒的修飾劑與保護劑。結構抑制是通過生物-離子液體獨特的空間位阻結構和多負電荷性質實現的。在微波輻射下能夠更好地提供熱量,並且快速地絡合在鐵粒子表面,隨著四氧化三鐵晶核逐漸形成,由於生物-離子液體存在獨特的空間位阻結構和多負電荷性質,在晶核表面形成了一個穩定保護層,阻礙鐵離子和亞鐵粒子與晶核的作用,進而抑制各晶面生長,來控制納米四氧化三鐵粒子尺寸和提高納米粒子的穩定性。得到的2-5nm的超小磁性納米簇具有優良的T1造影效果,具有臨床應用前景。(傳統方法及背景技術中的對比專利提到的8-100nm的磁珠具有的是T2造影效果,與本專利提到的超小納米簇T1效應是不一樣的)。
需要說明的是,只是施加微波,作用體為四氧化三鐵時,粒徑控制的不好,會出現粒徑不均一,且大於5nm,並且不具有T1造影效應。只有生物-離子液體的輔助作用下,才能很好地控制磁性納米簇的粒徑大小及分布,以及優良的T1造影效果。
與現有技術相比,本發明具有以下優點:
1)製備簡單、快捷、合成尺度大,只需使用少量幾種試劑即可製得。
2)微波輔助加熱具有反應速度快、節能、環保等優點。
3)可以直接製得水溶性良好的四氧化三鐵磁性納米簇,無需轉相等後續操作。
4)製得的四氧化三鐵磁性納米簇平均尺寸為2-5nm,且粒徑分布均勻。
5)表面攜帶羧基(-COOH)、氨基(-NH2)、羥基(-OH)等官能團,生物相容性好。
6)製得的四氧化三鐵磁性納米簇水溶液分散體穩定性好,靜置≧1年時未出現沉澱。
附圖說明
圖1為超小磁性納米簇的透射電鏡圖片。
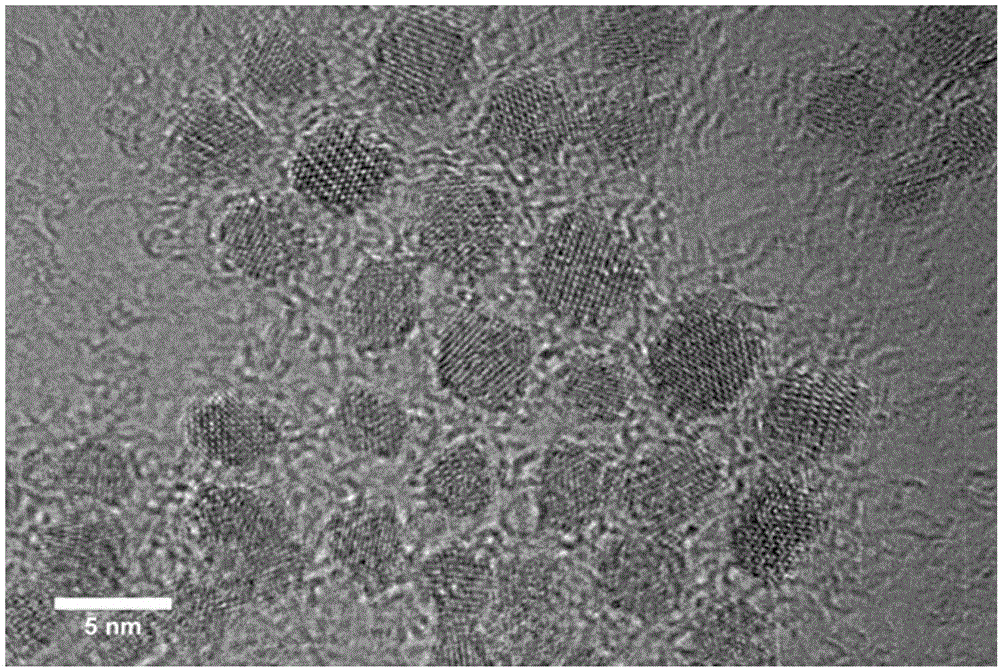
圖2為超小磁性納米簇的高分辨透射電鏡圖片。
圖3為超小磁性納米簇的X射線衍射圖。
圖4為超小磁性納米簇的傅立葉變換紅外光譜圖。
圖5為本發明的超小磁性納米簇在動物體內的T1核磁成像照片。
具體實施方式
下面結合附圖和具體實施例對本發明進行詳細說明。
實施例1
(1)稱取1mmol檸檬酸鐵和1mmol硫酸亞鐵,分別溶於10mL去離子水中,以200rpm的速度攪拌使其充分溶解。
(2)將步驟(1)製得的兩份溶液混合,繼續以200rpm的速度攪拌,使其充分混勻。
(3)稱取一定量的抗壞血酸,使其與Fe2+離子的摩爾比為1:3,加入步驟(2)得到的混合溶液中,繼續以200rpm的速度攪拌。
(4)向步驟(3)得到的混合溶液中加入0.1%的生物離子液體,所述的生物離子液體為膽汁素-檸檬酸鹽陰離子液體,繼續以200rpm的速度攪拌。
(5)向步驟(4)得到的混合溶液中滴加濃度為0.4M的氫氧化鈉溶液,調節該混合溶液的pH值至9,繼續以200rpm的速度攪拌5min。
(6)將步驟(5)得到的混合溶液轉移至微波反應罐中,所述的微波功率為500W,微波輻射頻率為2450MHz,反應溫度為100℃,反應時間為0.5h。
(7)將步驟(6)得到的產物轉移至透析袋中,透析1天,經差速離心,即可得到具有T1造影效應的四氧化三鐵磁性納米簇,所述的差速離心條件:離心速度10000rpm,離心時間2min。該磁性納米簇的r1弛豫率為3.5s-1mM-1,r2弛豫率為20s-1mM-1,r2/r1為5.71。
所製得的磁性納米簇的透射電鏡圖片、高分辨透射電鏡圖片、X射線衍射圖與傅立葉變換紅外光譜圖分別如圖1、圖2、圖3和圖4所示,從圖1和圖2可以看出,所述磁性納米簇呈均一的球體結構,粒徑大小為2-5nm。所得樣品經X射線衍射分析表明,所得產物的物相為Fe3O4(圖3)。同時,將所得樣品烘乾測定傅立葉變換紅外光譜,得知譜圖在1631cm-1出現羧酸跟反對稱伸縮振動峰及1385cm-1出現羧酸跟對稱伸縮振動峰,均屬於羧酸跟的特徵吸收峰,表明超小磁性納米簇表面含有羧基官能團(圖4)。
圖5為本實施例合成的超小磁性納米簇的T1核磁成像照片,通常的T2效應是灰度值越大,T2造影效應越強;而T1造影效應恰恰相反,即亮度值越大,T1造影效應越強。可知,本實施例的超小磁性納米簇具有優異的T1造影效果。
實施例2
(1)稱取1mmol乙醯丙酮鐵和2mmol氯化亞鐵,分別溶於17.5mL去離子水中,以350rpm的速度攪拌使其充分溶解。
(2)將步驟(1)製得的兩份溶液混合,繼續以350rpm的速度攪拌,使其充分混勻。
(3)稱取一定量的抗壞血酸,使其與Fe2+離子的摩爾比為1:12,加入步驟(2)得到的混合溶液中,繼續以350rpm的速度攪拌。
(4)向步驟(3)得到的混合溶液中加入0.3%的生物離子液體,所述的生物離子液體為膽汁素-馬來酸鹽陰離子液體,繼續以350rpm的速度攪拌。
(5)向步驟(4)得到的混合溶液中滴加濃度為2.7M的氫氧化鈉溶液,調節該混合溶液的pH值至10.5,繼續以200~500rpm的速度攪拌17.5min。
(6)將步驟(5)得到的混合溶液轉移至微波反應罐中,所述的微波功率為850W,微波輻射頻率為2450MHz,反應溫度為150℃,反應時間為2.5h。
(7)將步驟(6)得到的產物轉移至透析袋中,透析1.5天,經差速離心,即可得到具有T1造影效應的四氧化三鐵磁性納米簇,所述的差速離心條件:離心速度12000rpm,離心時間6min。該磁性納米簇的r1弛豫率為9s-1mM-1,r2弛豫率為35s-1mM-1,r2/r1為3.9。
實施例3
(1)稱取1mmol右旋糖酐鐵和3mmol硫酸亞鐵,分別溶於25mL去離子水中,以500rpm的速度攪拌使其充分溶解。
(2)將步驟(1)製得的兩份溶液混合,繼續以500rpm的速度攪拌,使其充分混勻。
(3)稱取一定量的抗壞血酸,使其與Fe2+離子的摩爾比為1:20,加入步驟(2)得到的混合溶液中,繼續以500rpm的速度攪拌。
(4)向步驟(3)得到的混合溶液中加入0.5%的生物離子液體,所述的生物離子液體為膽汁素-乙酸鹽陰離子液體,繼續以500rpm的速度攪拌。
(5)向步驟(4)得到的混合溶液中滴加濃度為5M的氫氧化鈉溶液,調節該混合溶液的pH值至12,繼續以500rpm的速度攪拌30min。
(6)將步驟(5)得到的混合溶液轉移至微波反應罐中,所述的微波功率為1200W,微波輻射頻率為2450MHz,反應溫度為200℃,反應時間為4h。
(7)將步驟(6)得到的產物轉移至透析袋中,透析2天,經差速離心,即可得到具有T1造影效應的四氧化三鐵磁性納米簇,所述的差速離心條件:離心速度14000rpm,離心時間10min。該磁性納米簇的r1弛豫率為3.5s-1mM-1,r2弛豫率為35s-1mM-1,r2/r1為10。
實施例4
(1)稱取1mmol檸檬酸鐵和4mmol氯化亞鐵,分別溶於10mL去離子水中,以500rpm的速度攪拌使其充分溶解。
(2)將步驟(1)製得的兩份溶液混合,繼續以500rpm的速度攪拌,使其充分混勻。
(3)稱取一定量的抗壞血酸,使其與Fe2+離子的摩爾比為1:12,加入步驟(2)得到的混合溶液中,繼續以500rpm的速度攪拌。
(4)向步驟(3)得到的混合溶液中加入1.5%的生物離子液體,所述的生物離子液體為膽汁素-琥珀酸鹽陰離子液體,繼續以500rpm的速度攪拌。
(5)向步驟(4)得到的混合溶液中滴加濃度為5M的氫氧化鈉溶液,調節該混合溶液的pH值至12,繼續以500rpm的速度攪拌5min。
(6)將步驟(5)得到的混合溶液轉移至微波反應罐中,所述的微波功率為300HZ,反應溫度為100℃,反應時間為3h。
(7)將步驟(6)得到的產物轉移至透析袋中,透析1天,經差速離心,即可得到具有T1造影效應的四氧化三鐵磁性納米簇,所述的差速離心條件:離心速度14000rpm,離心時間2min。該磁性納米簇的r1弛豫率為7.3s-1mM-1,r2弛豫率為27.5s-1mM-1,r2/r1為3.8。
實施例5
(1)稱取2mmol乙醯丙酮鐵和2mmol硫酸亞鐵,分別溶於25mL去離子水中,以200rpm的速度攪拌使其充分溶解。
(2)將步驟(1)製得的兩份溶液混合,繼續以200rpm的速度攪拌,使其充分混勻。
(3)稱取一定量的抗壞血酸,使其與Fe2+離子的摩爾比為1:8,加入步驟(2)得到的混合溶液中,繼續以200rpm的速度攪拌。
(4)向步驟(3)得到的混合溶液中加入0.5%的生物離子液體,所述的生物離子液體為NH2-PEG化咪唑二烴基磷酸鹽離子液體,繼續以200rpm的速度攪拌。
(5)向步驟(4)得到的混合溶液中滴加濃度為2M的氫氧化鈉溶液,調節該混合溶液的pH值至10,繼續以200rpm的速度攪拌30min。
(6)將步驟(5)得到的混合溶液轉移至微波反應罐中,所述的微波功率為500W,微波輻射頻率為2450MHz,反應溫度為200℃,反應時間為0.5h。
(7)將步驟(6)得到的產物轉移至透析袋中,透析1天,經差速離心,即可得到具有T1造影效應的四氧化三鐵磁性納米簇,所述的差速離心條件:離心速度10000rpm,離心時間10min。該磁性納米簇的r1弛豫率為9s-1mM-1,r2弛豫率為20s-1mM-1,r2/r1為0.45。
實施例6
(1)稱取1mmol右旋糖酐鐵和2mmol氯化亞鐵,分別溶於20mL去離子水中,以300rpm的速度攪拌使其充分溶解。
(2)將步驟(1)製得的兩份溶液混合,繼續以300rpm的速度攪拌,使其充分混勻。
(3)稱取一定量的抗壞血酸,使其與Fe2+離子的摩爾比為1:20,加入步驟(2)得到的混合溶液中,繼續以300rpm的速度攪拌。
(4)向步驟(3)得到的混合溶液中加入1%的生物離子液體,所述的生物離子液體為SH-PEG化咪唑二烴基磷酸鹽離子液體,繼續以300rpm的速度攪拌。
(5)向步驟(4)得到的混合溶液中滴加濃度為0.4M的氫氧化鈉溶液,調節該混合溶液的pH值至11,繼續以300rpm的速度攪拌20min。
(6)將步驟(5)得到的混合溶液轉移至微波反應罐中,所述的微波功率為1050W,微波輻射頻率為2450MHz,反應溫度為175℃,反應時間為3h。
(7)將步驟(6)得到的產物轉移至透析袋中,透析2天,經差速離心,即可得到具有T1造影效應的四氧化三鐵磁性納米簇,所述的差速離心條件:離心速度12000rpm,離心時間5min。該磁性納米簇的r1弛豫率為5s-1mM-1,r2弛豫率為23s-1mM-1,r2/r1為4.6。
實施例7
(1)稱取1mmol檸檬酸鐵、右旋糖酐鐵和2.5mmol硫酸亞鐵,分別溶於15mL去離子水中,以400rpm的速度攪拌使其充分溶解。
(2)將步驟(1)製得的兩份溶液混合,繼續以400rpm的速度攪拌,使其充分混勻。
(3)稱取一定量的抗壞血酸,使其與Fe2+離子的摩爾比為1:10,加入步驟(2)得到的混合溶液中,繼續以400rpm的速度攪拌。
(4)向步驟(3)得到的混合溶液中加入3%的生物離子液體,所述的生物離子液體為RCOOR-PEG化咪唑二烴基磷酸鹽離子液體,繼續以400rpm的速度攪拌。
(5)向步驟(4)得到的混合溶液中滴加濃度為4M的氫氧化鈉溶液,調節該混合溶液的pH值至10,繼續以400rpm的速度攪拌15min。
(6)將步驟(5)得到的混合溶液轉移至微波反應罐中,所述的微波功率為1100W,微波輻射頻率為2450MHz,反應溫度為100℃,反應時間為1h。
(7)將步驟(6)得到的產物轉移至透析袋中,透析1天,經差速離心,即可得到具有T1造影效應的四氧化三鐵磁性納米簇,所述的差速離心條件:離心速度13000rpm,離心時間5min。該磁性納米簇的r1弛豫率為8s-1mM-1,r2弛豫率為30s-1mM-1,r2/r1為3.75。
上述的對實施例的描述是為便於該技術領域的普通技術人員能理解和使用發明。熟悉本領域技術的人員顯然可以容易地對這些實施例做出各種修改,並把在此說明的一般原理應用到其他實施例中而不必經過創造性的勞動。因此,本發明不限於上述實施例,本領域技術人員根據本發明的揭示,不脫離本發明範疇所做出的改進和修改都應該在本發明的保護範圍之內。