槐定胺類衍生物及其製備方法和用途與流程
2023-09-23 10:46:05 1
本發明涉及醫藥領域,具體涉及槐定胺類衍生物及其製備方法和用途。
背景技術:
槐定鹼(sophoridine)是從豆科槐屬植物苦豆子中提取的生物鹼單體,其具有廣泛的藥理學作用,例如抗腫瘤、抗病毒、抗炎等。2005年鹽酸槐定鹼注射液被批准上市,用於治療滋養細胞癌。槐定鹼結構(結構如下所示)與現有抗癌化療藥結構不同,是一類新型的抗癌新藥,目前對槐定鹼結構改造及其構效關係研究較少,並且本領域對結構新穎且具有較高活性的抗腫瘤藥物仍有巨大需求。
技術實現要素:
本發明的發明人以槐定鹼為先導化合物,通過對槐定鹼的結構進行改造和修飾,得到了一類結構新穎的具有優異抗腫瘤活性的槐定胺類衍生物,本發明即是基於以上發現而完成。
本發明第一方面提供式i化合物、其光學異構體、其溶劑合物或其藥學上可接受的鹽,
其中,
r1和r2各自獨立地代表氫、c1-6烷基、c1-6烷氧基、芳基(例如6-20元芳基)、雜芳基(例如6-20元雜芳基)、環烷基(例如3-20元環烷基)或脂雜環基(3-20元脂雜環基),任選地,其中所述c1-6烷基、c1-6烷氧基、芳基、雜芳基、環烷基和脂雜環基被一個或多個(例如2、3、4、5或6個)選自下述的取代基取代:滷素、羥基、氨基、c1-6烷基、滷代c1-6烷基、c1-6烷氧基、3-8元環烷基、3-8元脂雜環基、6-15元芳基和5-15元雜芳基,其中,所述3-8元環烷基、3-8元脂雜環基、6-15元芳基和5-15元雜芳基任選地被一個或多個(例如1、2、3、4、5或6個)選自滷素、羥基、氨基、氰基、c1-6烷基、滷代c1-6烷基和c1-6烷氧基的取代基取代;或者,
r1和r2與相連的n原子一起形成脂雜環基(例如3-20元脂雜環基),其中所述脂雜環基任選地被一個或多個(例如1、2、3、4、5或6個)選自滷素、羥基、氨基、氰基、c1-6烷基、滷代c1-6烷基和c1-6烷氧基的取代基取代;
r3代表氫、滷素、羥基、氨基、氰基、c1-6烷基、滷代c1-6烷基和c1-6烷氧基;
n1代表0-10的整數(例如0、1、2、3、4、5、6、7、8、9或10);
a代表-ch2-、-co-或-s(o)n2-,其中n2為0、1或2;
環x代表芳環(例如6-20元芳環)或雜芳環(5-20元雜芳環);
代表碳氮單鍵或碳氮雙鍵,當代表碳氮雙鍵時,r1不存在。
在本發明的一個實施方案中,r1和r2各自獨立地代表氫、c1-6烷基、6-15元芳基(例如苯基、萘基)、苯並3-6元雜環基、3-8元脂雜環基,任選地,其中所述c1-6烷基、6-15元芳基、苯並3-6元雜環基、3-8元脂雜環基被一個或多個(例如1、2、3、4、5或6個)選自下述的取代基取代:滷素、羥基、氨基、氰基、c1-4烷基、滷代c1-4烷基、c1-4烷氧基和苯基,其中,所述c1-4烷基、c1-4烷氧基和苯基任選地被一個或多個(例如1、2、3、4、5或6個)選自滷素、羥基、氨基、三氟甲基、氰基、甲基和甲氧 基的取代基取代;或者,
r1和r2與相連的n原子一起形成3-8元脂雜環基,優選5-6元脂雜環基,例如吡咯烷基、吡咯烷酮基、咪唑烷基、哌啶基、哌嗪基、嗎啉基或硫嗎啉基,其中所述3-8元脂雜環基任選地被一個或多個(例如1、2、3、4、5或6個)選自滷素、羥基、氨基、三氟甲基、氰基、甲基和甲氧基的取代基取代;
代表碳氮單鍵或碳氮雙鍵,當代表碳氮雙鍵時,r1不存在。
在本發明的優選實施方案中,r1為氫,r2代表氫、c1-6烷基、6-15元芳基(例如苯基、萘基)、苯並3-6元雜環基、3-8元脂雜環基,任選地,其中所述c1-6烷基、6-15元芳基、苯並3-6元雜環基、3-8元脂雜環基被一個或多個(例如1、2、3、4、5或6個)選自下述的取代基取代:滷素、羥基、氨基、氰基、c1-4烷基、滷代c1-4烷基、c1-4烷氧基和苯基,其中,所述c1-4烷基、c1-4烷氧基和苯基任選地被一個或多個(例如1、2、3、4、5或6個)選自滷素、羥基、氨基、三氟甲基、氰基、甲基和甲氧基的取代基取代;或者,
r1和r2與相連的n原子一起形成3-8元脂雜環基,優選5-6元脂雜環基,例如吡咯烷基、吡咯烷酮基、咪唑烷基、哌啶基、哌嗪基、嗎啉基或硫嗎啉基,其中所述3-8元脂雜環基任選地被一個或多個(例如1、2、3、4、5或6個)選自滷素、羥基、氨基、三氟甲基、氰基、甲基和甲氧基的取代基取代;
代表碳氮單鍵。
在本發明的優選實施方案中,r1為氫,r2代表氫、c1-6烷基(優選甲基和乙基)、苯基、1,3-苯並二噁茂基或嗎啉基,任選地,其中所述c1-6烷基、苯基、1,3-苯並二噁茂基和嗎啉基被一個或多個(例如1、2、3、4、5或6個)選自下述的取代基取代:滷素、三氟甲基、甲氧基、1,3-苯並二噁茂基以及任選地被一個或多個(例如1、2、3、4、5或6個)選自滷素、三氟甲基、甲基和甲氧基的取代基取代的苯基;或者,
r1和r2與相連的n原子一起形成哌嗪環,其中所述哌嗪環任選地被 一個或多個(例如1、2、3、4、5或6個)選自滷素、三氟甲基、甲基和甲氧基的取代基取代;
代表碳氮單鍵。
在本發明的優選實施方案中,r1為氫,r2選自4-氯-3-三氟甲基苯基、3,4,5-三甲氧基苯基、3,4,5-三甲氧基苄基、4-嗎啉基、1,3-苯並二噁茂基、1,3-苯並二噁茂-4-甲基和1,3-苯並二噁茂-4-乙基;或者,
r1和r2與相連的n原子一起形成4-甲基-哌嗪基;
代表碳氮單鍵。
在本發明的一個實施方案中,r3代表氫、滷素(例如氟、氯、溴或碘)、羥基、氨基、氰基、c1-4烷基(例如甲基或以乙基)、滷代c1-4烷基或c1-4烷氧基(例如甲氧基或乙氧基),優選地,r3代表滷素,更優選地,r3為氯。
在本發明的一個實施方案中,環x為6-15元芳環或5-15元雜芳環,例如苯、萘、芴、吡咯、呋喃、噻吩、吡啶、吡嗪、嘧啶、吲哚或嘌呤,優選地,環x為苯。
在本發明的優選實施方案中,式i中為4-氯苯基。
在本發明的優選實施方案中,n1代表0-3的整數,更優選地,n1為3。
在本發明的優選實施方案中,a代表-ch2-、-co-或-so2-。
在本發明的一個實施方案中,所述式i化合物選自以下化合物,
12-n-對氯苄基-4』-(3,4,5-三甲氧基苯基)槐定胺;
12-n-對氯苄基-4』-(n-嗎啉基)槐定亞胺;
12-n-對氯苄基-4』-(2,3-亞甲二氧基苄基)槐定胺;
12-n-對氯苄基-4』-(3,4,5-三甲氧基苄基)槐定胺;
12-n-對氯苄基-4』-(4-氯-3-三氟甲基苯基)槐定胺;
12-n-對氯苄基-4』-(2,3-亞甲二氧基苯乙基)槐定胺;
12-n-對氯苄基-4』-(2,3-亞甲二氧基苯基)槐定胺;
12-n-對氯苄基-4』-(n-甲基哌嗪基)槐定胺;
12-n-對氯苯甲醯基-4』-(2,3-亞甲二氧基苄基)槐定胺;
12-n-對氯苯甲醯基-4』-(3,4,5-三甲氧基苄基)槐定胺;
12-n-對氯苯甲醯基-4』-(2,3-亞甲二氧基苯乙基)槐定胺;
12-n-對氯苯磺醯基-4』-(2,3-亞甲二氧基苄基)槐定胺;
12-n-對氯苯磺醯基-4』-(3,4,5-三甲氧基苄基)槐定胺;和
12-n-對氯苯磺醯基-4』-(2,3-亞甲二氧基苯乙基)槐定胺。
本發明式i化合物可通過以下方法製備得到:
方法一:
步驟一,槐定鹼經開環和酯化反應得到槐定酸甲酯z0a;
步驟二,z0a經n-烷基化得到z08;
步驟三,z08的酯基還原後得到c08;
步驟四,c08的羥基氧化後得到q08;
步驟五,q08的醛基與r1r2nh縮合(或經還原胺化反應)得到目標化合物target1;
方法二:
步驟1,化合物z0a的氨基經boc保護得到12-n-boc-槐定酸甲酯;
步驟2,12-n-boc-槐定酸甲酯還原後得到12-n-boc-槐定醇;
步驟3,12-n-boc-槐定醇氧化後得到12-n-boc-槐定醛;
步驟4,12-n-boc-槐定醛與r1r2nh經還原胺化反應得到12-n-boc-槐定胺衍生物;
步驟5,12-n-boc-槐定胺衍生物經脫保護反應得到目標化合物target2;
優選地,當r1r2nh中r1為氫時,在步驟5前還包括對12-n-boc-槐定胺衍生物中r2所連氨基進行選擇性保護的步驟,例如,選擇性保護基可以為fmoc;
方法三:
步驟a,z0a經n-磺醯化反應得到磺醯胺hxz08;
步驟b,hxz08的酯基經還原後得到hdc08;
步驟c,hdc08的醇經氧化後得到hxq08;
步驟d,hxq08與r1r2nh經還原胺化反應得到目標化合物target3。
本領域技術人員可通過教科書的教導製備得到上述各化合物,對於具體的反應條件本發明不作具體限定。
本發明的第二方面提供藥物組合物,其包含本發明第一方面任一項所述的式i化合物、其光學異構體、其溶劑合物或其藥學上可接受的鹽,以及任選地一種或多種藥學上可接受的載體或賦形劑。
本發明的第三方面提供本發明第一方面任一項所述的式i化合物、其光學異構體、其溶劑合物或其藥學上可接受的鹽在製備治療受試者所患癌症的藥物中的用途。
在本發明的一個實施方案中,所述癌症選自肝癌、肺腺癌、乳腺癌、胃癌、結腸癌、宮頸癌、卵巢癌、淋巴瘤神經膠質瘤、腦膠質瘤和黑色素瘤。
在本發明的一個實施方案中,所述受試者為哺乳動物,例如牛科動物、馬科動物、羊科動物、豬科動物、犬科動物、貓科動物、嚙齒類動物、靈長類動物;其中,特別優選的受試者為人。
本發明的另一方面提供一種治療癌症的方法,其包含向有需要的受試者施用有效量的本發明第一方面任一項所述的式i化合物、其光學異構體、其溶劑合物或其藥學上可接受的鹽。
在本發明的一個實施方案中,所述癌症選自肝癌、肺腺癌、乳腺癌、胃癌、結腸癌、宮頸癌、卵巢癌、淋巴瘤神經膠質瘤、腦膠質瘤和黑色素瘤。
在本發明的一個實施方案中,所述受試者為哺乳動物,例如牛科動物、馬科動物、羊科動物、豬科動物、犬科動物、貓科動物、嚙齒類動物、靈長類動物;其中,特別優選的受試者為人。
本發明的再一方面提供本發明第一方面任一項所述的式i化合物、其光學異構體、其溶劑合物或其藥學上可接受的鹽,其用於治療受試者所患的癌症。
在本發明的一個實施方案中,所述癌症選自肝癌、肺腺癌、乳腺癌、胃癌、結腸癌、宮頸癌、卵巢癌、淋巴瘤神經膠質瘤、腦膠質瘤和黑色素瘤。
在本發明的一個實施方案中,所述受試者為哺乳動物,例如牛科動物、馬科動物、羊科動物、豬科動物、犬科動物、貓科動物、嚙齒類動物、靈長類動物;其中,特別優選的受試者為人。
以下對本發明的術語進行解釋,對於特定的術語,如果本發明中的含義與本領域技術人員通常理解的含義不一致,以本發明中的含義為準;如果在本發明中沒有定義,則其具有本領域技術人員通常理解的含義。除非有相反陳述,本發明中使用的術語具有下述含義:
本發明所用術語「c1-6烷基」是指具有1-6(例如1、2、3、4、5或6)個碳原子的直鏈或支鏈烷基,例如c1-4烷基。所述c1-6烷基可被滷素取代得到所述滷代c1-6烷基,例如三氟甲基、三氟乙基等。具體的實例包括但不限於甲基、乙基、丙基、異丙基、正丁基、仲丁基、叔丁基、異丁基、戊基和己基等。
本發明所用術語「c1-6烷氧基」是指具有「c1-6烷基-o-」結構的基團,例如c1-4烷氧基,其中c1-6烷基具有與前文所述「c1-6烷基」相同的含義。具體的實例包括但不限於甲氧基、乙氧基、丙氧基、異丙氧基、正丁氧基、仲丁氧基、叔丁氧基、異丁氧基、戊氧基、2-戊氧基、3-戊氧基、異戊氧基、新戊氧基、仲戊氧基、己氧基、2-己氧基和3-己氧基等。
本發明中所用術語「環烷基」是指飽和或部分飽和的碳環,包括單環或多環,例如3-20元環烷基、3-15元環烷基、3-10元環烷基、3-8元環烷基、3-6元環烷基等。所述環烷基可任選地被一個或多個選自滷素、羥基、氨基、c1-6烷基、滷代c1-6烷基、c1-6烷氧基、3-8元環烷基、3-8元 脂雜環基、6-15元芳基和5-15元雜芳基的取代基取代。環烷基具體的實例包括但不限於環丙基、環丁基、環戊基和環己基等。
本發明所用術語「芳基」是指具有單環(如苯基)、多環(如萘基)或其中至少一個環是芳香性的稠合環(例如苯並3-6元雜環,如1,3-苯並二噁茂)的芳族碳環基,例如6-20元芳基、6-15元芳基等。所述芳基可任選地被一個或多個選自滷素、羥基、氨基、c1-6烷基、滷代c1-6烷基、c1-6烷氧基、3-8元環烷基、3-8元脂雜環基、6-15元芳基和5-15元雜芳基的取代基取代。芳基或取代芳基的具實例包括但不限於苯基、萘基、蒽基、菲基、芴基、茚基、苊基、1,3-苯並二噁茂、2-氯苯基、3-氯苯基、4-氯苯基、2-甲基苯基、3-甲基苯基、4-甲基苯基、2-甲氧基苯基、3-甲氧基苯基、4-甲氧基苯基、3,4,5-三甲氧基苯基、4-氯-3-三氟甲基苯基等。
本發明中所用術語「雜環基」是指含有至少一個、至多四個選自n、o或s的雜原子的單環或多環,可分為脂雜環基和芳雜環基,其中所述脂雜環基包括例如3-20元脂雜環基、3-15元脂雜環基、3-10元脂雜環基、3-8(例如3、4、5、6、7或8)元脂雜環基、3-6元脂雜環基等,其中所述芳雜環基(也稱作雜芳基)包括例如5-20元雜芳基,5-15元雜芳基、5-10元雜芳基或5-8元雜芳基等。所述雜環基可任選地被一個或多個選自滷素、羥基、氨基、c1-6烷基、滷代c1-6烷基、c1-6烷氧基、3-8元環烷基、3-8元脂雜環基、6-15元芳基和5-15元雜芳基的取代基取代。具體的實例包括但不限於環氧乙烷基、氧代環丁烷基、吡咯烷基、四氫呋喃基、二氫呋喃基、四氫噻吩基、吡咯基、呋喃基、噻吩基、咪唑基、噁唑基、噻唑基、哌啶基、四氫吡喃基、嗎啉基、硫嗎啉基、哌嗪基、4-甲基哌嗪基、吡啶基、吡喃基、吡嗪基、嘧啶基、三嗪基、吲哚基或嘌呤基等。
本發明所用術語「滷素」是指氟、氯、溴以及碘。
本發明所用術語「一個或多個」是指一個或兩個以上,例如1、2、3、4、5、6、7、8、9或10個。
本發明所用術語「光學異構體」包括本發明式1化合物的所有可能的光學異構體形式(例如對映異構體、非對映異構體等)。
本發明式1化合物或其藥學可接受的鹽還可以形成溶劑化物,例如水合 物、醇合物等,一般來說,與藥學可接受的溶劑如水、乙醇等形成的溶劑合物形式與非溶劑合物形式相當。
本發明式i化合物還可以是前藥或可在體內代謝變化後釋放出所述活性成分的形式。選擇和製備適當的前藥衍生物是本領域技術人員公知技術。
本文所用術語「有效量」是指足以實現所需治療效果的量,例如,實現減輕與待治療疾病相關的症狀的量。
本文所用的術語「治療」目的是減輕或消除所針對的疾病狀態或病症。如果受試者按照本文所述方法接受了治療量的化合物、其光學異構體或其藥學上可接受的鹽或其藥物組合物,該受試者一種或多種指徵和症狀表現出可觀察到的和/或可檢測出的降低或改善,則受試者被成功地「治療」了。還應當理解,所述的疾病狀態或病症的治療的不僅包括完全地治療,還包括未達到完全地治療,但實現了一些生物學或醫學相關的結果。
本文所用術語「藥物組合物」表示含有一種或多種本文所述化合物、其光學異構體或其藥學上可接受的鹽,以及藥學可接受的載體或賦形劑。藥物組合物的目的是促進對生物體的給藥,利於活性成分的吸收進而發揮生物活性。這裡所述的載體包括但不限於:離子交換劑,氧化鋁,硬脂酸鋁,卵磷脂,血清蛋白如人血白蛋白,緩衝物質如磷酸鹽,甘油,山梨酸,山梨酸鉀,飽和植物脂肪酸的部分甘油酯混合物,水,鹽或電解質,如硫酸魚精蛋白,磷酸氫二鈉,磷酸氫鉀,氯化鈉,鋅鹽,膠態氧化矽,三矽酸鎂,聚乙烯吡咯烷酮,纖維素物質,聚乙二醇,羧甲基纖維素鈉,聚丙烯酸酯,蜂蠟,羊毛脂。所述賦形劑是指在藥物製劑中除主藥以外的附加物。其性質穩定,與主藥無配伍禁忌,不產生副作用,不影響療效,在常溫下不易變形、乾裂、黴變、蟲蛀、對人體無害、無生理作用,不與主藥產生化學或物理作用,不影響主藥的含量測定等。如片劑中的黏合劑、填充劑、崩解劑、潤滑劑;中藥丸劑中的酒、醋、藥汁等;半固體製劑軟膏劑、霜劑中的基質部分;液體製劑中的防腐劑、抗氧劑、矯味劑、芳香劑、助溶劑、乳化劑、增溶劑、滲透壓調節劑、著色劑等均可稱為賦形劑。
本發明的化合物、其光學異構體、其溶劑合物或其藥學上可接受的鹽可以通過以下途徑給藥:胃腸外、局部、靜脈內、口服、皮下、動脈內、真 皮內、經皮、直腸、顱內、腹膜內、鼻內、肌內途徑或作為吸入劑。所述組合物可以任選地與在治療各種疾病中至少有一定效果的其它試劑聯合給藥。
本發明的化合物、其光學異構體或其藥學上可接受的鹽可根據給藥途徑配成各種適宜的劑型。
當口服用藥時,本發明化合物可製成任意口服可接受的製劑形式,包括但不限於片劑、膠囊、水溶液或水懸浮液。其中,片劑使用的載體一般包括乳糖和玉米澱粉,另外也可加入潤滑劑如硬脂酸鎂。膠囊製劑使用的稀釋劑一般包括乳糖和乾燥玉米澱粉。水懸浮液製劑則通常是將活性成分與適宜的乳化劑和懸浮劑混合使用。任選地,以上口服製劑形式中還可加入一些甜味劑、芳香劑或著色劑。
當皮膚局部施用時,本發明化合物可製成適當的軟膏、洗劑或霜劑製劑形式,其中將活性成分懸浮或溶解於一種或多種載體中。軟膏製劑可使用的載體包括但不限於:礦物油、液體凡士林、白凡士林、丙二醇、聚氧化乙烯、聚氧化丙烯、乳化蠟和水;洗劑或霜劑可使用的載體包括但不限於:礦物油、脫水山梨糖醇單硬脂酸酯、吐溫60、十六烷酯蠟、十六碳烯芳醇、2-辛基十二烷醇、苄醇和水。
本發明化合物還可以無菌注射製劑形式用藥,包括無菌注射水或油懸浮液或無菌注射溶液。其中,可使用的載體和溶劑包括水、林格氏溶液和等滲氯化鈉溶液。另外,滅菌的非揮髮油也可用作溶劑或懸浮介質,如單甘油酯或二甘油酯。
本發明的藥物製劑包括藥學上可實施的任何製劑,例如口服製劑、腸胃外給藥製劑等。
在本發明的實施方案中,進行合適的體外或體內測定來確定本發明藥物組合物的效果以及給藥是否適用於治療個體所患疾病或醫學疾病狀態。這些測定的實例在下文非限制性實施例結合具體疾病或醫學治療進行了描述。通常,足以實現預防或治療效果的本發明組合物的有效量為約0.001mg/千克體重/天至約10,000mg/千克體重/天。合適的情況下,劑量為約0.01mg/千克體重/天至約1000mg/千克體重/天。劑量範圍可以為每天、每兩天或每三天約0.01至1000mg/kg宿主體重,更通常為0.1至500mg/kg宿主體重。示例性 的治療方案為每兩天一次或每周一次或每月一次給藥。通常多次給予所述試劑,單次劑量之間的間隔可以是每天、每周、每月或每年。或者,可以以緩釋製劑的形式給予所述試劑,在這種情況下,需要較少的給藥頻率。劑量和頻率根據試劑在受試者中的半衰期而不同。也可以根據是預防性處理還是治療性處理而不同。在預防性應用中,以相對低頻率的間隔長期給予相對低的劑量。在治療性應用中,有時需要以相對短的間隔給予相對高的劑量,直至疾病的進展被延緩或停止,並優選地直至個體表現出疾病症狀的部分或完全改善,在此之後,可以給予患者預防方案。
發明的有益效果
本發明通過對槐定鹼11位和12位進行改造修飾得到一類結構新穎的具有優異抗腫瘤活性的化合物,例如對肝癌、肺腺癌、乳腺癌、胃癌、結腸癌、宮頸癌、卵巢癌、淋巴瘤、神經膠質瘤、腦膠質瘤和黑色素瘤具有較好的活性。在本發明的一個實施方案中,本發明的化合物對肝癌具有與一線抗腫瘤藥物(例如拓撲替康或紫杉醇)相當甚至更好的治療效果。
附圖說明
圖1為腹腔給藥後,溶劑對照組、多柔比星組和待測化合物組中裸鼠瘤體積變化曲線;
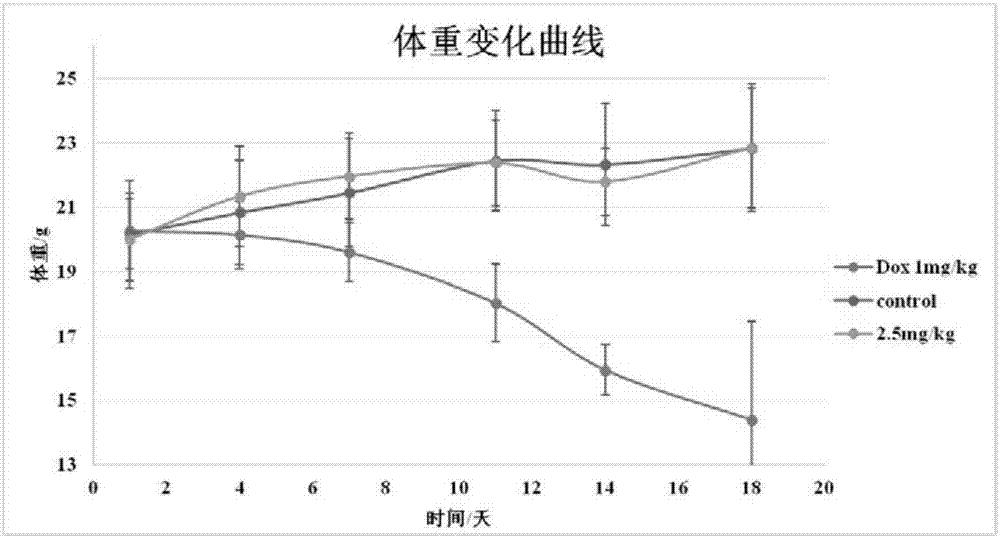
圖2為腹腔給藥後,溶劑對照組、多柔比星組和待測化合物組中裸鼠瘤體重變化曲線。
具體實施方式
下面將結合實施例對本發明的實施方案進行詳細描述,但是本領域技術人員將會理解,下列實施例僅用於說明本發明,而不應視為限定本發明的範圍。實施例中未註明具體條件者,按照常規條件或製造商建議的條件進行。所用試劑或儀器未註明生產廠商者,均為可以通過市購獲得的常規產品。
一、本發明化合物的合成
實施例112-n-對氯苄基槐定醛(q08)的合成
步驟一:將槐定鹼(4.96g,20mmol)均勻分散於50ml6nhcl水溶液中,回流反應4小時,tlc檢測原料消失後,停止反應,減壓蒸乾溶劑得黃色油狀物,加入50ml無水甲醇,室溫攪拌兩小時,tlc檢測反應完全,減壓蒸乾溶劑,得白色固體,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得到白色固體z0a。
步驟二:將上述白色固體z0a(2.81g,10.0mmol)懸浮於50ml1,2-二氯乙烷中,加入三乙胺(2.8ml,20.0mmol),攪拌至溶解,加入對氯苯甲醛(2.1g,15.0mmol),回流2小時後,緩慢加入三乙醯基硼氫化鈉(3.16g,15.0mmol),繼續回流至tlc檢測反應完全,冷至室溫後,依次用20ml水,20ml飽和食鹽水洗滌,有機層蒸乾後,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得白色固體z08。
步驟三:取四氫鋁鋰(0.5g,12.0mmol)懸浮於50ml無水四氫呋喃中,將z08(1.60g,4.0mmol)的四氫呋喃溶液15ml,緩慢加入上述體系中,室溫反應至tlc檢測原料消失,冰浴條件下依次緩慢加入0.5ml水,0.5ml15%naoh水溶液1.5ml淬滅反應體系,室溫攪拌0.5小時後過濾,減壓蒸乾濾液後,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色油狀物,加入10ml3n鹽酸乙醚溶液酸化過濾得黃色固體c08。
步驟四:氮氣保護下,向乾燥250ml三頸瓶中加入100ml無水二氯甲烷,冷至-78℃,依次緩慢滴加草醯氯(1.0ml,12mmol)和二甲基亞碸(1.5ml,20mmol),低溫攪拌5分鐘後,將c08(3.7g,10mmol)的二氯甲烷溶液 20ml,緩慢注射到上述反應體系中,繼續在-78℃反應半小時後,加入三乙胺(5.6ml,40mmol),室溫攪拌半小時後,依次用50ml水,50ml飽和食鹽水洗滌,減壓蒸乾有機相,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色粘稠固體12-n-對氯苄基槐定醛q08。
實施例212-n-對氯苄基-4』-(3,4,5-三甲氧基苯基)槐定胺(a08a)的合成
將q08(1.1g,3.0mmol)溶於50ml無水甲醇中,加入3,4,5-三甲氧基苯胺(0.8g,4.5mmol),回流4小時後,緩慢加入氰基硼氫化鈉(0.9g,4.5mmol)後,繼續回流4小時,冷至室溫後,減壓蒸乾反應體系,加入50ml二氯甲烷後,依次用30ml水,30ml飽和氯化銨溶液洗滌,減壓蒸乾有機相,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得白色固體(1.1g,67%)。熔點:77-79℃;1hnmr(500mhz,dmso-d6)δ7.43–7.25(m,4h),5.86(s,2h),3.70(s,6h),3.62–3.54(m,1h),3.53(s,3h),3.50–3.44(m,1h),3.09–2.63(m,6h),2.50–2.37(m,3h),2.30–2.12(m,1h),2.13–1.84(m,3h),1.81–1.60(m,3h),1.61–1.43(m,6h),1.44–1.34(m,1h),1.30–1.11(m,3h),1.11–0.88(m,1h);13cnmr(125mhz,dmso-d6)δ153.9,146.3(2),139.7(2),131.5,130.4,130.2,128.9(2),128.5(2),90.3,64.1,60.6,58.6(2),57.7,56.0,54.2,51.1,45.4,43.6,37.8,30.2,29.5,26.9,25.5,23.1,23.1,19.1;hrms:calcdforc31h44n3o3cl[m+h]+,542.3144,found,542.3147.
實施例312-n-對氯苄基-4』-(n-嗎啉基)槐定亞胺(a08b)的合成
將q08(1.1g,3.0mmol)溶於50ml無水甲醇中,加入4-氨基嗎啉(0.43ml,4.5mmol),回流4小時後,冷至室溫,減壓蒸乾反應體系,加入50ml二氯甲烷後,依次用30ml水,30ml飽和氯化銨溶液洗滌,減壓蒸乾有機相,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得白色固體(1.0g, 74%)。熔點:59-61℃;1hnmr(500mhz,dmso-d6)δ7.38–7.30(m,4h),6.93(t,j=5.2hz,1h),3.73–3.65(m,4h),3.61–3.44(m,2h),3.01–2.89(m,1h),2.88–2.75(m,6h),2.71–2.60(m,1h),2.50–2.35(m,2h),2.25–1.87(m,6h),1.82–1.62(m,3h),1.58–1.37(m,5h),1.31–1.21(m,2h),1.17–1.10(m,1h),1.01(dd,j=20.5,11.7hz,1h);13cnmr(125mhz,dmso-d6)δ140.4,139.7,131.4,130.2(2),128.5(2),66.1(2),63.9,58.5,57.7,54.4,52.5(2),52.4,51.6,45.4,38.0,32.9,29.8,26.8,26.1,25.5,22.6,19.2;hrms:calcdforc26h39n4ocl[m+h]+,459.2885;found,459.2885.
實施例412-n-對氯苄基-4』-(2,3-亞甲二氧基苄基)槐定胺(a08c)的合成
參考實施例3中方法,將q08與2,3,-亞甲二氧基苄胺按該條件反應,並做相應的後處理,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色油狀物,加入10ml3n鹽酸乙醚溶液酸化過濾得白色固體(收率:66%)。熔點:62-64℃;1hnmr(500mhz,dmso-d6)δ11.55(s,1h),11.14(s,1h),9.36(s,1h),9.25(s,1h),8.17–7.87(m,2h),7.70–7.47(m,2h),6.97–6.79(m,2h),6.78–6.62(m,1h),5.99(s,2h),4.51–4.34(m,1h),4.34–4.04(m,1h),3.92–3.71(m,1h),3.59(s,2h),3.41–3.11(m,5h),3.11–2.75(m,6h),2.76–2.55(m,2h),2.04–1.59(m,7h),1.56–1.17(m,4h),1.01–0.76(m,1h);13cnmr(125mhz,dmso-d6)δ147.8,146.4,133.1,131.5,129.4(2),129.2(2),122.1,109.5,108.8,101.3,61.0,56.4,55.5,55.0,52.0,48.4,46.7,44.6,33.9,31.5,26.0,25.3,23.1,22.3,22.0,21.7,19.0,17.6;hrms:calcdforc30h40n3o2cl[m+h]+,510.2881,found,510.2883.
實施例512-n-對氯苄基-4』-(3,4,5-三甲氧基苄基)槐定胺鹽酸鹽(a08d)的合成
參考實施例3中方法,將q08與3,4,5-三甲氧基苄胺按該條件反應,並做相應的後處理,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色油狀物,加入10ml3n鹽酸乙醚溶液酸化過濾得白色固體(收率:71%)。熔點:65-67℃;1hnmr(500mhz,dmso-d6)δ11.59(s,1h),11.17(s,1h),9.68(s,1h),9.57(s,1h),7.97(d,j=8.2hz,2h),7.53(d,j=8.3hz,2h),7.08(s,2h),4.50–4.19(m,2h),4.12–3.89(m,5h),3.80(s,6h),3.67(s,3h),3.32–2.93(m,6h),2.93–2.79(m,2h),2.79–2.54(m,2h),2.03–1.55(m,9h),1.60–1.19(m,3h),1.00–0.74(m,1h);13cnmr(125mhz,dmso-d6)δ153.2(2),138.2,134.7(2),133.1(2),129.4,129.2,127.9,108.3(2),65.3,61.1,60.4,56.6,55.5,55.0,52.0,50.6,46.3,44.6,33.9,26.0,25.3,25.2,23.2,22.3,22.1,21.7,17.6,15.6;hrms:calcdforc32h46n3o3cl.3hcl[m-3hcl+h]+,556.3300;found,556.3306.
實施例612-n-對氯苄基-4』-(4-氯-3-三氟甲基苯基)槐定胺(a08e)的合成
參考實施例3中方法,將q08與4-氯-3-三氟甲基苯胺按該條件反應,並做相應的後處理,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色油狀物,加入10ml3n鹽酸乙醚溶液酸化過濾得白色固體(收率:68%)。熔點:71-73℃;1hnmr(500mhz,dmso-d6)δ11.55(s,1h),11.12(s,1h),10.93(s,1h),8.12–7.77(m,2h),7.01(d,j=8.7hz,3h),6.91–6.74(m,2h),4.52–4.34(m,1h),4.34–4.12(m,1h),3.88–3.69(m,1h),3.65–3.25(m,4h),3.24–2.76(m,3h),2.76–2.55(m,3h),2.43–1.98(m,2h),1.99–1.29(m,8h),1.29–1.14(m,2h),1.03–0.79(m,1h);13cnmr(125mhz,dmso-d6)δ153.9,146.3,139.7,131.5,130.4,130.2,128.9(2),128.5(2),90.3,64.1,60.6,58.6,57.7,56.0,54.2,51.5,45.4,43.6,37.8,30.2,29.5,26.9,25.5,25.4,23.1,23.1,19.1;hrms:calcdforc29h36n3cl2f3[m+h]+,554.2311,found,554.2313.
實施例712-n-對氯苄基-4』-(2,3-亞甲二氧基苯乙基)槐定胺鹽酸鹽(a08f)的合成
參考實施例3中方法,將q08與2,3-亞甲二氧基苯乙胺按該條件反應,並做相應的後處理,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色油狀物,加入10ml3n鹽酸乙醚溶液酸化過濾得白色固體(收率:68%)。熔點:71-73℃;1hnmr(500mhz,dmso)δ11.56(s,1h),11.15(s,1h),9.56(s,1h),9.47(s,1h),7.97(d,j=8.2hz,2h),7.53(d,j=8.4hz,2h),7.29(d,j=1.3hz,1h),7.08(dd,j=8.0,1.4hz,1h),6.96(d,j=7.9hz,1h),6.06(s,2h),4.48–4.12(m,5h),4.10–3.91(m,2h),3.87–3.69(m,1h),3.33–2.92(m,6h),2.92–2.77(m,2h),2.76–2.55(m,3h),2.01–1.56(m,9h),1.54–1.20(m,3h),0.95–0.77(m,1h);13cnmr(125mhz,dmso-d6)δ148.1,147.7,133.1,129.4,129.2,125.9(2),124.6(2),110.9,108.6,101.7,65.3,63.3,61.1,55.5,55.0,52.0,50.0,49.0,46.0,44.6,33.8,26.0,25.2,23.2,22.3,22.1,21.7,17.6,15.6;hrms:calcdforc31h42n3o2cl.3hcl[m-3hcl+h]+,524.3038;found,524.3032.
實施例812-n-對氯苄基-4』-(2,3-亞甲二氧基苯基)槐定胺鹽酸鹽(a08g)的合成
參考實施例3中方法,將q08與2,3-亞甲二氧基苯胺按該條件反應,並做相應的後處理,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色油狀物,加入10ml3n鹽酸乙醚溶液酸化過濾得白色固體(收率:71%)。熔點:72-74℃;1hnmr(500mhz,dmso-d6)δ11.47(s,1h),11.46(s,1h),11.31(s,1h),11.06(s,1h),7.95(d,j=8.0hz,2h),7.52(d,j=8.3hz,2h),7.31–7.22(m,1h),7.14(d,j=8.2hz,1h),7.06(d,j=8.3hz,1h),6.09(s,2h),4.38(d,j=11.3hz,1h),4.31–4.22(m,1h),3.78(d,j=11.1hz,1h),3.34–3.02(m,7h),3.01–2.77(m,2h),2.74–2.66(m,1h),2.64–2.53(m,1h),2.50–2.35(m,1h),2.03–1.56(m,9h),1.54–1.23(m,3h),1.00–0.75(m,1h);13cnmr(125mhz,dmso-d6)δ148.3,147.7,134.7,133.1,129.4(2),129.2(2),116.9,109.0,104.6,102.5,65.3,63.3,61.2,55.5,55.0,52.05,50.8,49.0,44.6,33.9,26.1,25.1,23.0,22.3,22.1,21.7,17.6;hrms:calcdforc29h38n3o2cl.3hcl[m-3hcl+h]+,496.2725,found,496.2723.
實施例912-n-對氯苄基-4』-(n-甲基哌嗪基)槐定胺鹽酸鹽(a08h)的合成
參考實施例3中方法,將q08與n-甲基哌嗪按該條件反應,並做相應的後處理,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色油狀物,加入10ml3n鹽酸乙醚溶液酸化過濾得白色固體(收率:65%)。熔點:73-75℃;1hnmr(500mhz,dmso-d6)δ12.25(s,1h),12.15(s,1h),11.61(s,1h),11.16(s,1h),7.99(d,j=7.9hz,2h),7.57(d,j=7.9hz,2h),5.07(s,3h),4.50–4.16(m,1h),3.92–3.68(m,3h),3.66–3.35(m,5h),3.34–2.75(m,11h),2.73–2.53(m,2h),2.21–1.58(m,9h),1.60–1.23(m,3h),1.16–0.99(m,1h),1.01–0.80(m,1h);13cnmr(125mhz,dmso-d6)δ134.7,133.1,129.5(2),129.2(2),65.3,60.9,55.5,55.0,52.1(2),51.9(2),49.9,48.5,44.6,42.5,26.1,25.1,23.0,22.8,22.3,21.9,21.7,17.6,15.6;hrms:calcdforc27h43n4cl.4hcl[m-4hcl+h]+,459.3249;found,459.3249.
實施例1012-n-對氯苯甲醯基-4』-(2,3-亞甲二氧基苄基)槐定胺鹽酸鹽(xa08c)的合成
可參考如下合成路線:
步驟1:將z0a(2.81g,10.0mmol)懸浮於50ml二氯甲烷中,加入碳酸鉀粉末(1.4ml,10.0mmol),攪拌至溶解,加入二碳酸二叔丁酯(2.3ml,15.0mmol),室溫反應至tlc檢測反應完全,依次用50ml水、50ml飽和食鹽水洗滌後,無水硫酸鈉乾燥後過濾,減壓蒸乾,得12-n-boc-槐定酸甲酯粗品(2.53g)。
步驟2:將12-n-boc-槐定酸甲酯溶於100ml無水thf中,冰浴下緩慢加入lialh4(0.50g,12.5mmol),室溫反應至tlc檢測原料消失,冰浴下依次緩慢加入0.5ml水,0.5ml15%naoh水溶液1.5ml淬滅反應體系,室溫攪拌0.5小時後過濾,減壓蒸乾後加二氯甲烷溶解,依次用50ml水、50ml飽和食鹽水洗滌,無水硫酸鈉乾燥後過濾,減壓蒸乾,得12-n-boc-槐定醇粗品(2.17g)。
步驟3:氮氣保護下,向乾燥250ml三頸瓶中加入100ml無水二氯甲烷,冷至-78℃,依次緩慢滴加草醯氯(1.0ml,12mmol)和二甲基亞碸(1.5ml,20mmol),低溫攪拌5分鐘後,將12-n-boc-槐定醇(3.5g,10mmol)的二氯甲烷溶液20ml緩慢注射到上述反應體系中,繼續在-78℃反應半小時後,加入三乙胺(5.6ml,40mmol),室溫攪拌半小時後,依次用50ml水,50ml飽和食鹽水洗滌,減壓蒸乾有機相,得12-n-boc-槐定醛粗品3.3g。
步驟4:將12-n-boc-槐定醛(0.35g,1.0mmol)溶於50ml無水甲醇中, 加入2,3-亞甲二氧基苄胺(0.2ml,1.5mmol),回流4小時後,緩慢加入氰基硼氫化鈉(03g,1.5mmol)後,繼續回流4小時,冷至室溫後,減壓蒸乾反應體系,加入50ml二氯甲烷後,依次用20ml水,20ml飽和氯化銨溶液洗滌,減壓蒸乾有機相,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得12-n-boc-槐定胺(0.15g,43%)。
步驟5:將10ml12-n-boc-槐定胺(0.15g,0.43mmol)的二氧六環溶液加到20ml10%naco3水溶液中,冰浴下緩慢加入氯甲酸-9-芴基甲酯,室溫反應至tlc檢測原料消失,50ml二氯甲烷提取兩次後,減壓蒸乾得黃色粘稠狀固體。
步驟6:將步驟三中所得的固體溶於30ml二氯甲烷中,加入10ml三氟乙酸,室溫反應至tlc檢測原料消失,ph調至7-8,水洗後,無水硫酸鈉乾燥,過濾後,加入對氯苯甲醯氯(170ul,1.5mmol),室溫反應至tlc檢測原料消失,依次用10ml水,10ml飽和食鹽水洗滌,減壓蒸乾後,加入10ml哌啶,室溫攪拌1h,減壓蒸乾,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色油狀物,加入10ml3n鹽酸乙醚溶液酸化過濾得白色固體(123mg,23%)。熔點:87-89℃;1hnmr(500mhz,dmso-d6)δ11.26(s,1h),9.39(s,1h),9.33(s,1h),7.53–7.44(m,3h),7.42–7.31(m,1h),7.29–7.18(m,1h),7.08–7.00(m,1h),7.01–6.89(m,1h),6.05(s,2h),4.09–3.96(m,2h),3.68–3.20(m,8h),3.23–3.03(m,2h),3.01–2.73(m,2h),2.45–2.31(m,1h),1.88–1.51(m,8h),1.43–1.34(m,1h),1.38–1.08(m,4h);13cnmr(125mhz,dmso-d6)δ169.4,148.1,131.6,129.2,129.1,129.0,125.9(2),124.6(2),110.8,108.7,101.7,63.6,58.4,58.3,53.6,50.0,46.3,44.4,43.0,40.9,36.6,29.1,28.1,25.6,25.5,23.0,22.6,22.5,22.2,17.8;hrms:calcdforc30h38n3o3cl.3hcl[m-3hcl+h]+,524.2674;found,524.2674.
實施例1112-n-對氯苯甲醯基-4』-(3,4,5-三甲氧基苄基)槐定胺(xa08d)的合成
參考實施例10的方法,將步驟4中的2,3-亞甲二氧基苄胺替換為3,4,5- 三甲氧基苄胺,並做相應的後處理,終產品以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得白色固體(收率:71%)。熔點:72-74℃;1hnmr(500mhz,dmso-d6)δ7.65–7.17(m,4h),7.05–6.76(m,2h),5.77(s,1h),3.67(s,6h),3.57–3.38(m,4h),3.37–3.20(m,3h),3.17(s,3h),3.14–2.78(m,5h),2.44–2.06(m,3h),1.98–1.50(m,7h),1.47–1.29(m,3h),1.27–1.07(m,3h);13cnmr(125mhz,dmso-d6)δ169.3(2),153.7(2),135.5,135.4,134.5,134.3,129.2,129.1(2),129.0(2),60.5,58.4,56.6,53.7,52.2,49.0,46.5,44.4,40.8,36.6,30.0,29.2,28.1,27.3,25.6,23.39,23.0,22.2,17.8.
實施例1212-n-對氯苯甲醯基-4』-(2,3-亞甲二氧基苯乙基)槐定胺(xa08f)的合成
參考實施例10的方法,將步驟4中的2,3-亞甲二氧基苄胺替換為2,3-亞甲二氧基苯乙胺按該條件反應,並做相應的後處理,終產品以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得白色固體(收率:71%)。熔點:72-74℃;1hnmr(500mhz,dmso)δ7.58–7.25(m,4h),6.96–6.80(m,2h),6.77–6.59(m,1h),5.99(s,2h),3.77–3.56(m,2h),3.54–3.32(m,6h),3.21–2.77(m,11h),2.44–2.07(m,1h),2.03–1.48(m,7h),1.47–1.04(m,4h);13cnmr(125mhz,dmso-d6)δ169.4,147.8,146.4,135.4,134.5,131.5(2),129.2(2),122.1,109.4,108.8,101.3,72.6,66.8,60.5,58.4,53.6,49.03,48.3,46.9,39.0,36.6,31.5,29.4,28.17,25.1,25.8,23.6,22.4,17.8;hrms:calcdforc31h40n3o3cl[m+h]+,538.2831;found,538.2830.
實施例1312-n-對氯苯磺醯基槐定醛(hxq08)的合成
步驟a:將z0a(2.81g,10.0mmol)懸浮於50ml二氯甲烷中,加入碳酸鉀粉末(1.4ml,10.0mmol),攪拌至溶解,加入對氯苯磺醯氯(2.6g,15.0mmol),室溫反應8小時,tlc檢測原料消失,依次用20ml水,20ml飽和食鹽水洗滌,有機層蒸乾後,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得白色固體hxz08。
步驟b:取四氫鋁鋰(0.5g,12.0mmol)懸浮於50ml無水四氫呋喃中,將hdz08(1.82g,4.0mmol)的四氫呋喃溶液15ml,緩慢加入上述體系中,室溫反應至tlc檢測原料消失,冰浴條件下依次緩慢加入0.5ml水,0.5ml15%naoh水溶液1.5ml淬滅反應體系,室溫攪拌0.5小時後過濾,減壓蒸乾濾液後,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色油狀物hdc08。
步驟c:氮氣保護下,向乾燥250ml三頸瓶中加入100ml無水二氯甲烷,冷至-78℃,依次緩慢滴加草醯氯(1.0ml,12mmol)和二甲基亞碸(1.5ml,20mmol),低溫攪拌5分鐘後,將hdc08(4.2g,10mmol)的二氯甲烷溶液20ml,緩慢注射到上述反應體系中,繼續在-78℃反應半小時後,加入三乙胺(5.6ml,40mmol),室溫攪拌半小時後,依次用50ml水,50ml飽和食鹽水洗滌,減壓蒸乾有機相,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得黃色粘稠固體12-n-對氯苯磺醯基槐定醛hxq08。
實施例1412-n-對氯苯磺醯基-4』-(2,3-亞甲二氧基苄基)槐定胺(hxa08c)的合成
將hxq08(1.2g,3.0mmol)溶於50ml無水甲醇中,加入2,3-亞甲二氧基苄胺(0.6ml,4.5mmol),回流4小時後,緩慢加入氰基硼氫化鈉(0.9g,4.5mmol)後,繼續回流4小時,冷至室溫後,減壓蒸乾反應體系,加入50ml二氯甲烷後,依次用30ml水,30ml飽和氯化銨溶液洗滌,減壓蒸乾有機相,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得白色固體(1.0g,61%)。熔點:87-89℃;1hnmr(500mhz,dmso-d6)δ7.87–7.74(m,2h),7.71–7.60(m,2h),7.11–7.01(m,1h),6.96–6.84(m,2h),6.02(s,2h),3.89–3.73(m,2h),3.73–3.62(m,1h),3.53–3.45(m,1h),2.97–2.71(m,4h),2.60–2.51(m,2h),2.47–2.33(m,1h),2.03–1.91(m,1h),1.79–1.67(m,2h),1.66–1.34(m,6h),1.33–0.98(m,8h);13cnmr(125mhz,dmso-d6)δ147.7,147.2,140.4,137.7,129.9(2),128.9(2),123.1,109.8,108.5,101.4,59.3,57.6,53.9,51.4,47.3,45.1,45.0,37.6,29.3,28.4,27.3,26.6,25.3,24.4,24.2,18.7,15.6;hrms:calcdforc29h38n3o4cls[m+h]+,560.2344;found,560.2372.
實施例1512-n-對氯苯磺醯基-4』-(3,4,5-三甲氧基苄基)槐定胺(hxa08d)的合成
參考實施例14的方法,將hxq08與3,4,5-三甲氧基苄胺按該條件反應,並做相應的後處理,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得白色固體(收率:65%)。熔點:83-85℃;1hnmr(400mhz,dmso-d6)δ7.88–7.68(m,2h),7.69–7.53(m,2h),6.68(s,2h),3.76(s,6h),3.69–3.64(m,3h),3.63(s,3h),3.52–3.27(m,2h),2.84–2.62(m,4h),2.32(d,j=10.5hz,1h),2.05–1.83(m,1h),1.81–1.48(m,5h),1.48–0.97(m,12h);13cnmr(100mhz,dmso-d6)δ153.1(2),140.5,137.7,136.6,135.8,129.8(2),128.9(2),105.7(2),60.4,59.5,57.6,56.2(2),54.2,53.2,48.62,45.3,45.1,38.0,29.6,29.0,28.9,26.4,25.8,24.8,24.6,18.9;hrms:calcdforc31h44n3o5cls[m+h]+, 606.2763;found,606.2786.
實施例1612-n-對氯苯磺醯基-4』-(2,3-亞甲二氧基苯乙基)槐定胺(hxa08f)的合成
參考實施例14的方法,將hxq08與2,3-亞甲二氧基苯乙胺按該條件反應,並做相應的後處理,以二氯甲烷/甲醇為流動相,經矽膠柱層析分離純化,得白色固體(收率:67%)。熔點:82-84℃;1hnmr(500mhz,dmso)δ7.81(d,j=8.6hz,2h),7.67(d,j=8.6hz,2h),6.91–6.79(m,2h),6.74–6.61(m,1h),5.98(s,2h),3.66(t,j=7.2hz,1h),3.53–3.44(m,1h),2.96–2.86(m,2h),2.86–2.70(m,6h),2.63(t,j=7.4hz,2h),2.39–2.25(m,1h),2.03–1.82(m,1h),1.80–1.51(m,5h),1.51–1.37(m,3h),1.33–1.14(m,6h),1.12–0.95(m,3h);13cnmr(125mhz,dmso-d6)δ147.7,146.1,140.5,137.7,129.9(2),128.9(2),122.0,109.4,108.6,101.2,65.4,59.4,57.6,54.1,49.6,47.9,45.2,45.0,37.8,33.2,29.3,27.1,26.5,25.6,24.7,24.2,18.8,15.6;hrms:calcdforc30h40n3o4cls[m+h]+,574.2500;found,574.2526.
實驗例1本發明化合物對體外細胞的抑制活性測定(ic50)
待測化合物:本申請實施例化合物;
拓撲替康:購於中國藥品食品檢定院;
紫杉醇:購於中國藥品食品檢定院;
用dmso將待測化合物配置成30mg/ml溶液,加入pbs配置成所需的五個梯度濃度,在96孔板上以4﹡103的密度接種處於對數生長期的hepg2細胞(購於中國科學院上海生命科學研究院),貼壁生長24h後加藥,每個化合物設3個復孔,並設dmso溶媒對照孔和無細胞調零孔。於37℃、5%co2條件下培養48h,每孔加入20μl5mg/mlmtt溶液,繼續培養4h,吸出孔內培養液後,每孔加入dmso液150μl,將培養板置於微孔板振蕩器上振蕩10分鐘,酶標儀測定各孔光吸收,測定波長為492nm。根據各孔od值計算藥物對細胞增殖抑制率。計算公式為抑制率(%)=(1-化合物od492/對 照od492)×100。將各濃度梯度的抑制率數據導入sigmaplot軟體,計算ic50值。
表1槐定胺類衍生物抗肝癌hepg2細胞活性結果
實驗例2本發明化合物的體內藥效評價
實驗用雌性balb/c小鼠(購於中國醫學科學院實驗動物研究所),將1×107人肝癌細胞hepg2細胞(購於中國科學院上海生命科學研究院)用pbs配置成0.2ml細胞懸液,無菌條件下在動物右側腋窩皮下接種腫瘤。接種後三周脫頸處死,剝離腫瘤表層組織,剪碎成2mm×2mm瘤塊,接種於18-22g實驗用balb/c裸鼠右側腋窩皮下,每組8隻共3組:陽性藥物對照組(dox,多柔比星(購於中國藥品食品檢定院))、溶劑對照組(生理鹽水)和待測化合物組(化合物a08c)。接種腫瘤後72小時,a08c與多柔比星用0.9%生理鹽水溶解,分別配置成0.25mg/ml和0.1mg/ml的溶液,腹腔給藥量為 0.2ml/20g,對照組注射生理鹽水,每四天稱量體重,測量瘤塊直徑並計算瘤體積(瘤體積=1/2×長徑×短徑2)。接種後18天結束實驗,稱體重,處死動物,剝取腫瘤並稱重。實驗結果見圖1和圖2。
儘管本發明的具體實施方式已經得到詳細的描述,本領域技術人員應當理解,根據已經公開的所有教導,可以對本發明的細節進行各種修改和替換,這些改變均在本發明的保護範圍之內。本發明的全部範圍由所附權利要求及其任何等同物給出。