2、2′-亞甲基-雙[4-特辛基-6-(2H‑苯並三唑基-2)]苯酚的無溶劑合成的製作方法
2023-09-20 23:50:10 3
本發明涉及的是一種2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚的無溶劑合成方法,具體地說是先以二正丙基胺和甲醛或多聚甲醛為原料合成二(正丙基氨基)甲烷,再和2-(2′-羥基-5′-特辛基苯基)苯並三唑在無溶劑的條件下通過縮合合成2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚,屬於精細化工及高分子材料助劑領域。
背景技術:
2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚是一種性能優異的苯並三唑類紫外線吸收劑,又名紫外線吸收劑UV-360,2,2′-亞甲基雙(4-叔辛基-6-苯並三唑苯酚)或2、2′-亞甲基雙[6-(2H-苯並三唑基-2-基)-4-(1,1,3,3-四甲基丁基)苯酚]等,適用於PP、PE,也可用於聚苯乙烯、ABS樹脂、PVC、尼龍、聚氨酯等。
UV-360的合成方法較多,但有工業價值的合成方法主要是先用甲醛、二烷基胺和一分子的2-(2′-羥基-5′-特辛基苯基)苯並三唑(又稱UV-329)反應,生成2-(二烷基氨基)甲基-6-(苯並三唑基-2)苯酚,然後,在鹼的催化作用下,在高沸點芳烴類溶劑中與第二分子的UV-329反應,生成UV-360(梁松傑.一種受阻胺類光穩定劑的製備方法[P].CN 104592136A,2015-05-06;Kubota N,Nishimura A.Process for preparing 2、2′-methylene-bis-(4-hydrocarbyl-6-benzo-triazolylphenols)[P].EP0180993A2,1986-05-14)。該方法的反應式如下:
其中,R1,R2=乙基,正丙基,異丙基等。
該方法也可採用一鍋法合成(張曉東,勾少萍;範小鵬等.2、2′-亞甲基雙[6-(2H-苯並三唑基-2-基)-4-(1,1,3,3-四甲基丁基)苯酚]的製備方法[P].CN 105566238A,2016-05-11;Leistner W E,Moschchitisky S,Levent M.Process for preparing 2、2′-methylene-bis(6-(2H-benzo-triazol-2-yl)-4-hydrocarbylphenol s)[P].US5237071,1993-08-17。儘管以上方法具有收率較高的優點,但在第二步縮合反應中均要用二甲苯、三甲苯、異丙基苯等沸點比較高的有機溶劑作為反應介質,這些溶劑與副產物二烷基胺及產品分離比較困難,影響二烷基胺及溶劑的循環使用,殘存在產品中還影響產品的質量。
技術實現要素:
為了克服以上方法存在的問題,本發明的發明者對合成2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚進行了深入的研究,發現先以二正丙基氨和甲醛(或多聚甲醛)為原料,通過縮合合成雙(二正丙基氨基)甲烷,然後在鹼的催化作用下,在無溶劑的條件下和UV-329縮合可順利地合成2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚。該方法的合成原理如下:
由於不用高沸點溶劑,克服了現有方法存在的問題,即二正丙基氨容易回收及循環使用,產品質量高。
本發明的技術方案為:
一種2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚的無溶劑合成方法,其工藝步驟如下:
(1)雙(二正丙基氨基)甲烷的合成:在裝有攪拌器、溫度計和油水分離器的反應瓶中加入二正丙基胺和37%的甲醛水溶液(或多聚甲醛),在不斷攪拌下,加熱到120℃進行脫水縮合反應,反應至無水蒸出(約3h)時,結束反應。先常壓下蒸出過量的二正丙基胺,再減壓蒸出雙(二正丙基氨基)甲烷(100-110℃/20mmHg)。
(2)UV-360的合成:在裝有攪拌器、溫度計和蒸餾接收裝置的反應瓶中加入UV-329、粉末狀的氫氧化鈉和雙(二正丙基氨基)甲烷,在攪拌下加熱到120℃,使物料全部融化,升高反應溫度至140℃,並反應1h後,減壓(100mmHg)蒸出反應生成的二正丙基胺,再提高反應溫度到160℃,並於減壓(100mmHg)下反應2~3h。停止加熱,並排除負壓。
(3)UV-360的提純:待溫度降至110℃時慢慢加入甲苯,使物料全部溶解。物料溶解後,使溫度降到90℃,慢慢加入質量百分數為18%的鹽酸,攪拌,使物料充分中和至中性。將反應物轉移到燒杯中,用冰水冷卻結晶4~5h後,過濾,濾餅先用水洗滌兩次,再用甲醇洗滌兩次,並於100~110℃烘乾得UV-360。
進一步地,所述的二正丙基胺和37%的甲醛水溶液的質量比為1:0.35~0.50,優選為1:0.36~0.40,或二正丙基胺和多聚甲醛的質量比為1:0.14~0.18,優選為1:0.15~0.16。
所述的UV-329和雙(二正丙基氨基)甲烷的質量比為1:0.65~1:0.80,優選為1:0.70~1:0.75。
所述的UV-329和氫氧化鈉的質量比為1:0.11~1:0.14,優選為1:0.12~1:0.13。
所述的產品提純過程中所用甲苯的質量為UV-329的1.3~1.7倍,優選為1.5~1.6倍。
所述的產品提純過程中所用甲醇的質量為UV-329的1.3~1.7倍,優選為1.5~1.6倍。
本發明的一種2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚的無溶劑合成方法的優點在於:不用高沸點溶劑,有利於二正丙基胺的回收及循環使用,並且反應時間短、產品收率和質量高。
附圖說明
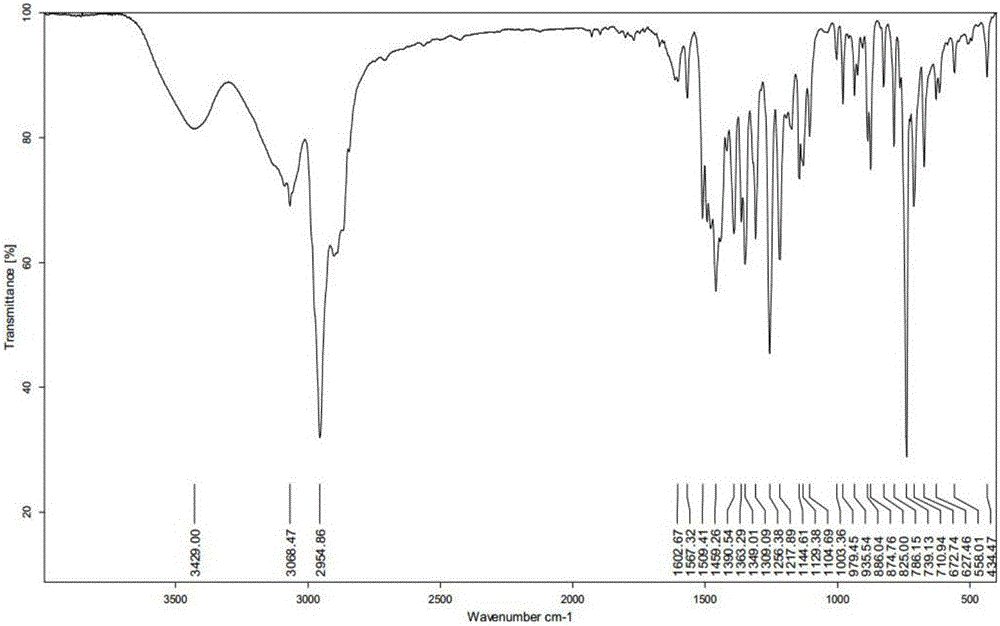
圖1為本發明實施例1得到的2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚的紅外光譜圖。
圖2為本發明實施例1得到的2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚的核磁共振氫譜圖。
圖3為本發明實施例1得到的2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚的核磁共振碳譜圖。
具體實施方式
以下對本發明的優選實施例進行說明,應當理解,此處所描述的優選實施例僅用於說明和解釋本發明,並不用於限定本發明。
除非另有說明,本發明中所採用的百分數均為質量百分數。
實施例中2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚的含量採用高效液相色譜分析。液相色譜分析條件為,色譜柱:HyperODS2C18柱(250mm×4.6mm);流動相:V(乙腈)/V(水)=90/10;流速:1.0mL/min;柱溫:室溫;檢測波長:210nm。所用液相色譜儀為美國沃特世公司的Waters 600型高效液相色譜儀。二正丙基胺和雙(二正丙基氨基)甲烷的含量採用氣相色譜分析,所用儀器為島津GC-14C氣相色譜儀,分析條件如下:柱型:AC1.10,進樣溫度200℃,檢測溫度:300℃,流動相:氯仿,採用程序升溫,每分鐘20℃。色譜數據採用浙大智能N2000數據工作站處理,含量採用面積歸一法計算。
實施例1
雙(二正丙基氨基)甲烷的合成,其工藝步驟如下:
在裝有攪拌器、溫度計和油水分離器的1000mL三口燒瓶中加入444g二正丙基胺和162g 37~40%的甲醛水溶液,在不斷攪拌下,加熱到120℃進行脫水縮合反應,反應至無水蒸出(約3h)時,結束反應。先常壓下蒸出過量的二正丙基胺約50g,再減壓蒸餾,收集100~110℃/20mmHg)餾分,得雙(二正丙基氨基)甲烷401g(理論428g),收率93.69%,含量98.8%。
實施例2
雙(二正丙基氨基)甲烷的合成,其工藝步驟如下:
在裝有攪拌器、溫度計和油水分離器的1000mL三口燒瓶中加入444g二正丙基胺和64g 94%的多聚甲醛,在不斷攪拌下,加熱到120℃進行脫水縮合反應,反應至無水蒸出(約2h)時,結束反應。先常壓下蒸出過量的二正丙基胺約50g,再減壓蒸餾,收集100~110℃/20mmHg)餾分,得雙(二正丙基氨基)甲烷402g(理論428g),收率93.93%,含量98.9%。
實施例3
2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚的無溶劑合成,其工藝步驟如下:
(1)UV-360的合成:在裝有攪拌器、溫度計和蒸餾接收裝置的反應瓶中加入323g UV-329、4g粉末狀的氫氧化鈉和128g雙(二正丙基氨基)甲烷,在攪拌下加熱到120℃,使物料全部融化,升高反應溫度至140℃,並反應1h後,減壓(100mmHg)蒸出反應生成的二正丙基胺,再提高反應溫度到160℃,並於減壓(100mmHg)下反應2h。停止加熱,並排除負壓。反應過程中回收二正丙基胺103g(理論量122g),回收率84.4%,含量98.7%。
(2)UV-360的提純:待溫度降至110℃時慢慢加入500g甲苯,使物料全部溶解。物料溶解後,使溫度降到90℃,慢慢加入質量百分數為18%的鹽酸(約40g),攪拌,使物料充分中和至中性。將反應物轉移到燒杯中,用冰水冷卻結晶4~5h後,過濾,濾餅先用150mL×2的去離子水洗滌兩次,再用250g×2的甲醇洗滌兩次,並於100~110℃烘乾得UV-360 231g(理論產量329g),產品熔點197~198℃,產率70.2%,含量99.3%。
本發明還通過紅外和核磁共振測定對本實施例得到的產物結構進行了表徵。圖1為本發明實施例1得到的UV-360的紅外光譜圖;圖2為本發明實施例1得到的UV-360的1H NMR(500MHz,CDCl3);圖3為本發明實施例1得到的UV-360的13C NMR(126MHz,CDCl3)。
圖1中,3429cm-1為酚羥基的O-H伸縮振動峰,1256cm-1為酚羥基的O-H彎曲振動峰,2955cm-1為飽和碳上的C-H伸縮振動峰,1459cm-1為飽和碳上的C-H彎曲振動峰,1391和1363cm-1為叔丁基上的C-H彎曲振動峰,3066cm-1為芳環上的C-H伸縮振動峰,1603和1567cm-1為芳環上的C=C伸縮振動峰,875和739為芳環上的C-H彎曲振動峰。以上分析說明圖1中的紅外光譜符合UV-360的分子結構特徵。
圖2中,0.72ppm處的峰為9號位置(甲基)的氫質子峰,1.41ppm處的峰為7號位置(甲基)的氫質子峰,1.75ppm處的峰為8號位置(亞甲基)的氫質子峰,4.32ppm處的峰為4號位置(亞甲基)的氫質子峰,7.38ppm處的峰為6號位置(苯環)的氫質子峰,7.47ppm處的峰為2號位置(苯環)的氫質子峰,7.94ppm處的峰為1號位置(苯環)的氫質子峰,8.31ppm處的峰為5號位置(苯環)的氫質子峰,11.54ppm處的峰為3號位置(酚羥基)的氫質子峰。以上分析說明圖2中氫核的化學位移符合UV-360的分子結構特徵。
圖3中,145.71ppm處的峰為3號位置(甲基)的碳核峰,142.76ppm處的峰為9號位置的碳核峰,141.36ppm處的峰為5號位置的碳核峰,129.93ppm處的峰為4號位置的碳核峰,129.40ppm處的峰為2號位置的碳核峰,127.4ppm處的峰為6號位置的碳核峰,124.51ppm處的峰為1號位置的碳核峰,117.61ppm處的峰為8號位置的碳核峰,116.72ppm處的峰為10號位置的碳核峰,56.68ppm處的峰為13號位置的碳核峰,38.22ppm處的峰為11號位置的碳核峰,32.33ppm處的峰為14號位置的碳核峰,32.07ppm處的峰為15號位置的碳核峰,30.90ppm處的峰為12號位置的碳核峰,32.33ppm處的峰為7號位置的碳核峰,76.75ppm、77.00ppm、77.26ppm處的峰為溶劑CHCl3中碳核的三重峰。以上分析說明圖2中碳核的化學位移符合UV-360的分子結構特徵。
以上分析表明本實施例合成的產物為UV-360。
實施例4
2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚的無溶劑合成,其工藝步驟如下:
(1)UV-360的合成:在裝有攪拌器、溫度計和蒸餾接收裝置的反應瓶中加入323g UV-329、4g粉末狀的氫氧化鈉和128g雙(二正丙基氨基)甲烷,在攪拌下加熱到120℃,使物料全部融化,升高反應溫度至140℃,並反應1h後,減壓(100mmHg)蒸出反應生成的二正丙基胺,再提高反應溫度到160℃,並於減壓(100mmHg)下反應3h。停止加熱,並排除負壓。反應過程中回收二正丙基胺103.5g(理論量122g),回收率84.8%,含量98.6%。
(2)UV-360的提純:待溫度降至110℃時慢慢加入500g甲苯,使物料全部溶解。物料溶解後,使溫度降到90℃,慢慢加入質量百分數為18%的鹽酸(約40g),攪拌,使物料充分中和至中性。將反應物轉移到燒杯中,用冰水冷卻結晶4~5h後,過濾,濾餅先用150mL×2的去離子水洗滌兩次,再用250g×2的甲醇洗滌兩次,並於100~110℃烘乾得UV-360 231.3g(理論產量329g),產品熔點197~198℃,產率70.3%,含量99.2%。
按照實施例3中的表徵方式對本實施例的產品進行檢測,證明本實施例得到的產物為目標產物。
實施例5
一種2、2′-亞甲基-雙[4-特辛基-6-(2H-苯並三唑基-2)]苯酚的無溶劑合成方法,其工藝步驟如下:
(1)雙(二正丙基氨基)甲烷的合成:在裝有攪拌器、溫度計和油水分離器的1000mL三口燒瓶中加入444g二正丙基胺(回收)和162g 37~40%的甲醛水溶液,在不斷攪拌下,加熱到120℃進行脫水縮合反應,反應至無水蒸出(約3h)時,結束反應。先常壓下蒸出過量的二正丙基胺約50g,再減壓蒸餾,收集100~110℃/20mmHg)餾分,得雙(二正丙基氨基)甲烷400g(理論428g),收率93.45%,含量98.6%。
(2)UV-360的合成:在裝有攪拌器、溫度計和蒸餾接收裝置的反應瓶中加入323g UV-329、4g粉末狀的氫氧化鈉和128g雙(二正丙基氨基)甲烷,在攪拌下加熱到120℃,使物料全部融化,升高反應溫度至140℃,並反應1h後,減壓(100mmHg)蒸出反應生成的二正丙基胺,再提高反應溫度到160℃,並於減壓(100mmHg)下反應2h。停止加熱,並排除負壓。反應過程中回收二正丙基胺103.2g(理論量122g),回收率84.6%,含量98.9%。
(3)UV-360的提純:待溫度降至110℃時慢慢加入500g甲苯,使物料全部溶解。物料溶解後,使溫度降到90℃,慢慢加入質量百分數為18%的鹽酸(約40g),攪拌,使物料充分中和至中性。將反應物轉移到燒杯中,用冰水冷卻結晶4~5h後,過濾,濾餅先用150mL×2的去離子水洗滌兩次,再用250g×2的甲醇洗滌兩次,並於100~110℃烘乾得UV-360 230.6g(理論產量329g),產品熔點197~198℃,產率70.1%,含量99.2%。
按照實施例3中的表徵方式對本實施例的產品進行檢測,證明本實施例得到的產物為目標產物。
最後應說明的是:以上所述僅為本發明的優選實施例而已,並不用於限制本發明,儘管參照前述實施例對本發明進行了詳細的說明,對於本領域的技術人員來說,其依然可以對前述各實施例所記載的技術方案進行修改,或者對其中部分技術特徵進行等同替換。凡在本發明的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發明的保護範圍之內。