一種利用乙基纖維素為碳源製備磷酸錳鐵鋰正極材料的方法與流程
2023-09-09 16:11:20 2
本發明涉及鋰離子電池正極材料製備領域,具體地指一種利用乙基纖維素為碳源製備磷酸錳鐵鋰正極材料的方法。
背景技術:
自1991年sony公司首次推出商品鋰離子電池以來,鋰離子電池已經以其開路電壓高、循環壽命長、能量密度高、自放電低、無記憶效應、對環境友好等優點廣泛應用於人們工作、學習、生活的各個方面。近年來,隨著動力電池及大型電力儲能裝置的市場需求不斷增加,陸續出現了以鋰離子電池作為載體的動力及儲能電源。
消費類電池用到的正極材料多數為LiCoO2,因其價格昂貴,具有毒性等缺點並不適合於對能量密度和安全性更高的動力電池應用領域。純電動汽車和混合動力汽車的蓬勃發展,帶動了動力電池的生產,同時也對動力電池的能量密度、安全性和成本等因素提出了更高的要求。橄欖石型正極材料LiFePO4的安全性好,成本低,循環性能好,滿足了動力電池的要求,但在能量密度上還存在不足。LiMnPO4與LiFePO4同屬橄欖石結構,具有同LiFePO4相同的比容量,更高的工作電壓(4.IV,磷酸鐵鋰為3.4V),更高的比能量(701Wh/Kg,磷酸鐵鋰為586Wh/Kg),更低廉的成本。然而相較於LiFePO4,LiMnPO4晶格內部阻力大,電子/離子傳導速率較慢,電導率小於10-10S/cm,比磷酸鐵鋰還要低兩個數量級以上。電子在LiFePO4中發生躍遷的能隙為0.3eV,有半導體特徵;而磷酸錳鋰的能隙為2eV,其電子導電性差,屬絕緣體。
為了提高磷酸錳鋰的電子導電率,人們一般採用碳包覆,金屬離子摻雜,材料納米化三種方式。碳包覆能夠有效的提高LiMnPO4顆粒的導電性。但是LiMnPO4表面包覆的碳是非活性物質,碳加入量過多不但會影響材料的振實密度和加工性能,同時在一定程度上減小了LiMnPO4與電解液的接觸面積,阻礙了Li+的運動。另外傳統的碳源不能完整均勻包覆在LiMnPO4顆粒表面,因此製備包碳量較少且具有較高電化學性能的LiMnPO4材料具有重要的生產意義。目前,在改善碳包覆工藝方面已做了大量工作。例如,中國專利(公布號CN103594712A,公布日2014.02.19)公開一種金屬摻雜的氧化導電碳黑包覆磷酸錳鋰及其製備方法,步驟是:先將磷酸二氫鋰、二氧化錳、磷酸、氫氧化鎂及氧化導電碳黑混合後分散於水中,經球磨後的漿料。得到的漿料噴霧乾燥後置於保護性氣氛中以2℃/min的升溫速率升溫至500℃。恆溫燒結12h,冷卻至室溫後粉碎、過篩,得化學式為LiMg0.05Mn0.95PO4/C的金屬摻雜的氧化導電碳黑包覆磷酸錳鋰。採用無機物作為碳源時,無機碳源的親水性差難以以層狀形式包覆在材料表面。首先影響材料的導電性其次正極材料中的活性物質與電解液直接接觸Mn離子溶解於電解液中。中國專利(公布號CN105513820A,公開日2016.04.20)公開一種碳包覆的磷酸錳鋰材料的製備方法,步驟是:將納米磷酸錳鋰材料、蔗糖和聚乙烯毗咯烷酮(PVP)以質量比為1:0.25:1分散在去離子水中,充分攪拌均勻,其中固含量為20%。經霧化烘乾處理,得到微米球形的碳包覆的磷酸錳鋰材料。霧化速率為5ml/min,烘乾條件為進料口溫度為220℃,出料口溫度為120℃。將出料口得到的粉體置於管式爐中,通以氫氣氣氛,從室溫以5℃/min的升溫速率到達600℃,鍛燒4h,自然冷卻至室溫。上述方法以蔗糖為碳源,最終複合物中的碳含量約為10%,碳源的添加量過多,雖然可以大大提高磷酸錳鋰的倍率性能,同時碳源添加量過多會導致合成產物的克容量降低,影響電池的電容量。
技術實現要素:
本發明針對當前技術中存在的LiMnPO4材料電子導電性差,材料表面碳包覆不均勻,充放電過程中極化嚴重,大倍率性能差,提出了採用乙基纖維素作為碳源,對正極材料進行包碳提高材料電化學性能。本發明利用乙基纖維素能很好的溶於有機溶劑並且碳化後質量小的特點,球磨後能完整均勻包覆在正極材料表面,燒結碳化後正極材料表面碳膜厚度小從而顯著提高材料電化學性能。
本發明技術方案為:
一種利用乙基纖維素為碳源製備磷酸錳鐵鋰正極材料的方法,包括以下步驟:
①按照製備的磷酸錳鐵鋰的原料的離子摩爾比來稱取錳源化合物、鐵源化合物、磷源化合物,然後將它們連同還原劑溶解於溶劑一中得到A液;將氫氧化鋰溶解於溶劑二中得到B液,然後將B液滴加至A液中得到磷酸錳鋰的前驅體溶液,將其置於高溫高壓反應釜中,加熱至160-300℃,反應時間為1-20h,待反應釜冷卻至室溫後洗滌、真空乾燥,製得磷酸錳鐵鋰前驅體材料;
其中,製備的磷酸錳鐵鋰的原料的離子摩爾比為鋰離子:錳離子:鐵離子:磷酸根離子=3-3.3:X:1-X:1,X=0.1-0.9;理論量磷酸錳鋰為按照磷酸根離子摩爾數全部得到磷酸錳鋰的質量;體積比為A液:B液=0.5-2:1;還原劑的加入量在前驅體溶液中的總濃度為0.03-0.2mol/L;前驅體溶液中鋰離子濃度為0.1-1mol/L;
②將步驟①所得的前驅體材料與碳源按質量比1-30:1,並以溶劑三為介質在球磨中混合,其中,每克碳源加入5-30ml的溶劑三,球磨轉速200-500r/min,時間為2-10h,得到混合物;所述碳源為乙基纖維素、羥乙基纖維素、羥丙基纖維素、甲基纖維素或纖維素衍生物;
③將步驟②所得混合物乾燥後放入管式爐中,在惰性氣體氛圍下,先在240-390℃條件下預燒1-5h,隨爐冷卻至室溫,然後將其研磨破碎,再放入管式爐中,同樣在惰性氣氛、450-780℃下,燒結3-12h,隨爐冷卻至室溫,最後得到碳包覆的磷酸錳鐵鋰正極材料。
所述的步驟①中溶劑一和溶劑二均為水與有機溶劑的混合物,體積比水:有機溶劑=0.1-1:1,有機溶劑為乙醇、乙二醇、丙三醇、聚乙二醇或聚丙烯酸。
所述的步驟①中前驅體真空乾燥條件40-60℃,-0.1MPa。
所述的步驟①中的還原劑為葡萄糖、抗壞血酸、草酸、乙酸和檸檬酸中的一種或多種。
所述的步驟②中的溶劑三為苯、甲苯、乙苯、二甲苯、無水乙醇、甲醇、乙醇、丙醇、乙二醇和丙三醇中的一種或多種。
所述的步驟③中的惰性氣氛為氮氣或氬氣。
所述的步驟①中B液滴加至A液中的滴加時間為15-20min。
所述的錳源為硫酸亞錳、硝酸亞錳、醋酸錳、檸檬酸錳和氯化亞錳中一種或多種。
所述的鐵源為硫酸亞鐵、硝酸亞鐵、草酸亞鐵和氯化鐵中的一種或幾種。
所述的前驅體溶液的體積為反應釜容積的30-90%。
最終產物中包碳量為磷酸錳鐵鋰質量的0.5-30%。
本發明的有益效果為:
本發明公開了一種利用乙基纖維素為碳源製備磷酸錳鐵鋰正極材料的方法。先水熱法製備磷酸錳鐵鋰前驅體,再與乙基纖維素在合適的有機溶劑中球磨包碳後經預燒、燒結得到磷酸錳鐵鋰包碳正極材料。當前技術中,水熱法大部分都是採用葡萄糖、蔗糖作為碳源,這類碳源在球磨包碳時如果採用水為球磨介質,但是糖遇水後的糖漿後續很難處理,所以採用醇類為球磨介質後續處理比較容易,但這又導致包碳不均勻,因為葡萄糖、蔗糖在醇中溶解性差。而採用乙基纖維素可以很好的解決以上問題因為它能在有機溶劑中溶解(溶脹)。
乙基纖維素是一種熱塑性、非離子型的纖維素烷基醚,是氯乙烷與鹼纖維素的反應產物。乙基纖維素是典型的疏水性藥用輔料,乙基纖維素能很好的溶於有機溶劑,在醫藥領域乙基纖維素常與增塑劑按一定比例混合配製成薄膜包衣液,本發明利用乙基纖維素能在材料表面很好的成膜特性。球磨時採用乙基纖維素與一定體積的有機溶液構成包衣液,球磨後乙基纖維素能完整均勻包覆在磷酸錳鐵鋰表面,燒結碳化後正極材料表面碳層均勻完整。
正極材料表面碳層較厚不利於電化學性能發揮,原因有二:首先碳材料是惰性的,另外碳層厚度較大會引起鋰離子通過碳薄膜的擴散距速率降低,限制內部活性物質的有效利用。從圖1可知乙基纖維素惰性氣氛下碳化後碳殘留較小,650℃殘留率為3.5%。這就導致燒結碳化後正極材料表面碳膜厚度小,利於鋰離子在活性物質與電解液之間的傳遞從而顯著提高材料電化學性能。
基於乙基纖維素在材料表面良好的成膜特性與碳化殘留率小的特點,燒結後正極材料表面碳層均勻完整且較薄,使材料能在較小的包碳量條件下獲得較高比容量,增大正極材料的體積能量密度,例如實施例1中採用乙基纖維素做為碳源,最終包碳量為2%時,0.1C下153.9mAh/g,5C下102.0mAh/g。其性能甚至優於相同製備方法採用葡萄糖作為碳源,最終包碳量為7%下材料0.1C下的149.7mAh/g,5C下的85.3mAh/g。從而具有極大的實際應用價值。
附圖說明
圖1為實施例1中採用的碳源-乙基纖維素與實施例5中採用的碳源-葡萄糖的的熱失重曲線圖;
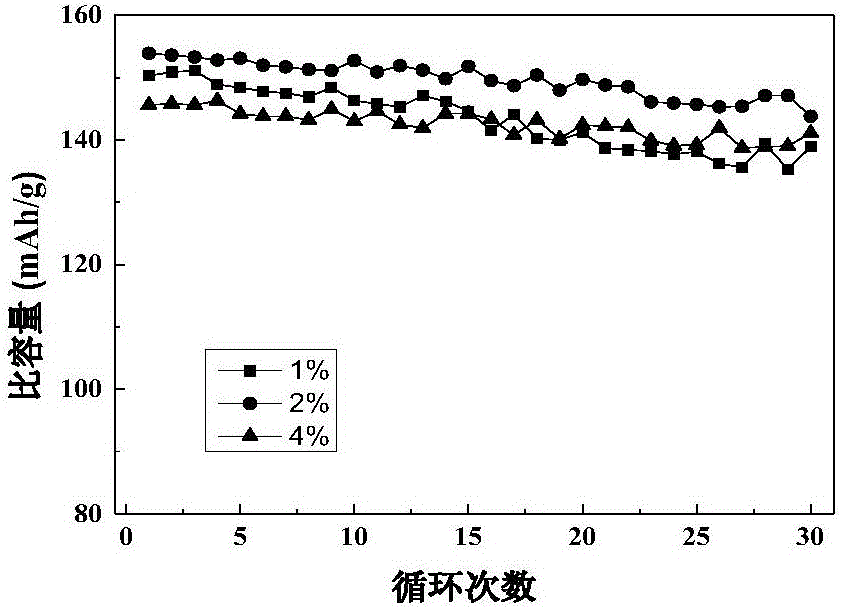
圖2為實施例1、4、5中採用不同乙基纖維素包覆量時磷酸錳鐵鋰循環充放電曲線圖;
圖3為實施例1中採用乙基纖維素作為碳源與實施例6、7中相同工藝條件下但採用葡萄糖作為碳源時磷酸錳鐵鋰倍率性能曲線圖;
圖4為實施例1中採用乙基纖維素為碳源時得到磷酸錳鐵鋰包碳材料的SEM圖;
圖5為實施例1中採用乙基纖維素為碳源時得到磷酸錳鐵鋰包碳材料的XRD譜圖。
具體實施方式:
下面結合附圖和實施例對本發明進一步說明。
水熱過程中的反應方程式為:(實施例1、2、4、5、6、7)
3LiOH+XMnSO4+(1-X)FeSO4+H3PO4→LiMnxFe1-xPO4+Li2SO4+3H2O
其中X=0.1-0.9;
實施例1:
①硫酸亞錳(0.0384mol)、硫酸亞鐵(0.0096mol)、磷酸(0.048mol)、抗壞血酸(0.014mol)(即按照LiMnXFe1-XPO4(X=0.8)進行稱量)溶解於160ml水與乙二醇按體積比=1:2組成的混合溶劑中,稱為A液;將氫氧化鋰(0.144mol)溶解於160ml水與乙二醇按體積比=1:2組成的混合溶劑中溶劑二中,稱為B液。然後將B液20min滴加至A液中得到磷酸錳鐵鋰的前驅體溶液,其中前驅體溶液中抗壞血酸的總濃度為0.0438mol/L,前驅體溶液中鋰離子濃度為0.45mol/L。將其置於高溫高壓反應釜中,前驅體溶液的體積為反應釜容積的80%,加熱至240℃,反應時間為4h,待反應釜冷卻至室溫後分別用蒸餾水、乙醇離心洗滌三次,50℃、-0.1MPa真空乾燥後製得磷酸錳鐵鋰前驅體材料;
②將步驟①所得的前驅體材料與乙基纖維素按質量比15:2,稱取前驅體材料3g,按每克乙基纖維素加入25ml無水乙醇為介質,球磨混合,球磨轉速300r/min,時間為6h。
③將步驟②所得混合物乾燥後放入管式爐中,在氮氣氣體氛圍下燒結,先在350℃條件下預燒3h,隨爐冷卻至室溫,然後將其研磨破碎,再加入管式爐中,在氮氣氣氛下燒結,燒結工藝分別為650℃,6h,隨爐冷卻至室溫,得到碳包覆的LiMn0.8Fe0.2PO4正極材料。最終產物中包碳量為LiMn0.8Fe0.2PO4質量的2%。(經元素分析儀測試得到正極材料中碳含量為2%)
圖1為利用同步熱分析儀在氮氣氣氛下以10℃/min升溫速率、溫度範圍25-800℃分別對乙基纖維素和葡萄糖熱失重情況的測試。可以看出本發明所採用的碳源乙基纖維素在氮氣氣氛下650℃最終碳殘留為3.6%比常用碳源葡萄糖的13.1%小。
另外由於乙基纖維素可以溶於乙醇,而葡萄糖在乙醇中微溶。所以採用乙基纖維素作為碳源最終正極材料表面的碳層均勻完整且較薄,可以有效地提高材料電化學性能。
圖2為利用CT2001A型LAND測試儀對電池進行充放電測試,電壓範圍為2.0-4.6V,測試溫度25℃。可以看出用在0.1C倍率下乙基纖維素作碳源材料的一致性都比較好;當乙基纖維素燒結後殘留碳佔磷酸錳鐵鋰2%時的材料電化學性能最好。
圖3為利用CT2001A型LAND測試儀對電池進行倍率性能測試,電壓範圍為2.0-4.6V,測試溫度25℃。可以看出當採用乙基纖維素為碳源包碳量為2%時,在0.1C下比容量達到153.9mAh/g,5C下比容量為102.0mAh/g,在不同倍率下30次循環充放電後在0.1C下比容量無衰減。但採用葡萄糖為碳源包碳量同為2%時,0.1C下比容量為142.3mAh/g,5C下比容量為50.6mAh/g,在不同倍率下30次循環充放電後在0.1C下比容量衰減嚴重。並且乙基纖維素為碳源包碳2%材料性能要優於以葡萄糖為碳源包碳7%材料性能。說明正極材料以乙基纖維素為碳源可以在較小的包碳量下獲得較高的電化學性能。
圖4為利用NanoSEM450型掃描電子顯微鏡對樣品進行形貌表徵,可以看出採用乙基纖維素為碳源製得的材料粒徑分布均勻,粒徑在200nm左右。
圖5為利用D8 Focus型X–射線衍射儀對樣品進行晶體結構表徵,測試條件為:Cu靶Kα(λ=0.154 06nm)射線,掃描速率為12°/min,步長為0.02°,範圍為10°-80°。可以看出,本方法製備出的磷酸錳鐵鋰的XRD譜圖與標準譜圖相吻合,且峰型尖銳無雜質峰出現,說明用本方法能夠製備出晶型完整的磷酸錳鐵鋰材料。
實施例2:
①硫酸亞錳(0.0096mol)、硫酸亞鐵(0.0384mol)、磷酸(0.048mol)、抗壞血酸(0.024mol)、葡萄糖(0.024mol)(即按照LiMnXFe1-XPO4(X=0.2)進行稱量)溶解於200ml水與乙二醇按體積比=1:1組成的混合溶劑中,稱為A液;將氫氧化鋰(0.1548mol)溶解於120ml水與乙二醇按體積比=1:1組成的混合溶劑中溶劑二中,稱為B液。然後將B液15min滴加至A液中得到磷酸錳鐵鋰的前驅體溶液,其中前驅體溶液中抗壞血酸、葡萄糖的總濃度為0.15mol/L,前驅體溶液中鋰離子濃度為0.4838mol/L。將其置於高溫高壓反應釜中,前驅體溶液的體積為反應釜容積的40%,加熱至200℃,反應時間為4h,待反應釜冷卻至室溫後分別用蒸餾水、乙醇離心洗滌三次,60℃、-0.1MPa真空乾燥後製得磷酸錳鐵鋰前驅體材料;
將步驟①所得的前驅體材料與乙基纖維素按質量比3:1,按每克乙基纖維素加入15ml丙醇為介質球磨混合,球磨轉速450r/min,時間為4h。
將步驟②所得混合物乾燥後放入管式爐中,在氬氣氣體氛圍下燒結,先在300℃條件下預燒4h,隨爐冷卻至室溫,然後將其研磨破碎,再加入管式爐中,在氬氣氣氛下燒結,燒結工藝分別為600℃,8h,隨爐冷卻至室溫,得到碳包覆的LiMn0.2Fe0.8PO4正極材料。最終產物中包碳量為LiMn0.2Fe0.8PO4質量的5%。
實施例3:
本實施例水熱過程中的反應方程式為:
3LiOH+XMn(NO3)2+(1-X)Fe(NO3)2+H3PO4→LiMnxFe1-xPO4+2LiNO3+3H2O
其中X=0.1-0.9;
具體實驗為:
①硝酸亞錳(0.0336mol)、硝酸亞鐵(0.0144mol)、磷酸(0.048mol)、抗壞血酸(0.0096mol)(即按照LiMnXFe1-XPO4(X=0.7)進行稱量)溶解於160ml水與乙二醇按體積比=1:2組成的混合溶劑中,稱為A液;將氫氧化鋰(0.144mol)溶解於120ml水與乙二醇按體積比=1:2組成的混合溶劑中溶劑二中,稱為B液。然後將B液20min滴加至A液中得到磷酸錳鐵鋰的前驅體溶液,其中前驅體溶液中抗壞血酸的總濃度為0.0343mol/L,前驅體溶液中鋰離子濃度0.5143mol/L。將其置於高溫高壓反應釜中,前驅體溶液的體積為反應釜容積的90%,加熱至180℃,反應時間為8h,待反應釜冷卻至室溫後分別用蒸餾水、乙醇離心洗滌三次,60℃、-0.1MPa真空乾燥後製得磷酸錳鐵鋰前驅體材料;
將步驟①所得的前驅體材料與羥丙基纖維素按質量比15:1,按每克羥丙基纖維素加入25ml乙苯為介質球磨混合,球磨轉速500r/min,時間為4h。
將步驟②所得混合物乾燥後放入管式爐中,在氮氣氣體氛圍下燒結,先在350℃條件下預燒4h,隨爐冷卻至室溫,然後將其研磨破碎,再加入管式爐中,同樣在氮氣氣氛下燒結,燒結工藝分別為650℃,6h,隨爐冷卻至室溫,得到碳包覆的LiMn0.7Fe0.3PO4正極材料。最終產物中包碳量為LiMn0.7Fe0.3PO4質量的1%。
實施例4:
其他步驟同實施例1,不同之處為步驟②中改為:將步驟①所得的前驅體材料與乙基纖維素按質量比15:4,稱前驅體材料3g。按每克乙基纖維素加入25ml無水乙醇為介質,球磨混合,球磨轉速300r/min,時間為6h。最終得到採用乙基纖維素為碳源的碳包覆量為4%的LiMn0.8Fe0.2PO4。
實施例5:
其他步驟同實施例1,不同之處為步驟②中改為:將步驟①所得的前驅體材料與乙基纖維素按質量比15:1,稱前驅體材料3g。按每克乙基纖維素加入25ml無水乙醇為介質,球磨混合,球磨轉速300r/min,時間為6h。最終得到採用乙基纖維素為碳源的碳包覆量為1%的LiMn0.8Fe0.2PO4。
實施例6:
其他步驟同實施例1,不同之處為步驟②中改為:將步驟①所得的前驅體材料與葡萄糖按質量比18:5,稱前驅體材料3g。按每克葡萄糖加入25ml無水乙醇為介質,球磨混合,球磨轉速300r/min,時間為6h。最終得到採用葡萄糖為碳源的碳包覆量為7%的LiMn0.8Fe0.2PO4。
實施例7:
其他步驟同實施例1,不同之處為步驟②中改為:將步驟①所得的前驅體材料與葡萄糖按質量比12:1,稱前驅體材料3g。按每克葡萄糖加入25ml無水乙醇為介質,球磨混合,球磨轉速300r/min,時間為6h。最終得到採用葡萄糖為碳源的碳包覆量為2%的LiMn0.8Fe0.2PO4。
本發明未盡事宜為公知技術。