運行氣相光氣化裝置的方法與流程
2023-10-31 11:26:32 7
大量製備異氰酸酯並主要用作製備聚氨酯的起始物質。它們通常通過使用化學計算過量的光氣使相應的胺與光氣反應製備。胺與光氣的反應可以在氣相也可以在液相中進行。在氣相中的工藝方案(通常被稱作氣相光氣化)的特徵在於選擇反應條件以使至少反應組分胺、異氰酸酯和光氣,優選所有反應物、產物和反應中間產物在所選條件下為氣態。氣相光氣化的優點尤其包括降低的光氣存在(Aufkommen)(所謂的光氣「滯留」)、避免難以光氣化的中間產物、提高的反應收率和較低能量要求,因為使用較少溶劑。本發明僅涉及氣相光氣化,尤其涉及啟動氣相光氣化裝置的順利方法。
現有技術公開了通過在氣相中使胺與光氣反應而製備異氰酸酯的各種方法。良好工藝方案的重要因素是氣相光氣化的反應物的良好混合。EP-A-0 289 840描述了通過氣相光氣化製備二異氰酸酯,其中根據該發明的製備在沒有活動件的柱形空間內在湍流中在200℃至600℃的溫度下進行。
EP-A-0 570 799 涉及製備芳族二異氰酸酯的方法,其中相應的二胺與光氣的反應在管式反應器中在高於二胺的沸點下在0.5至5秒的平均接觸時間內進行。
EP-A-0 699 657描述在氣相中製備芳族二異氰酸酯的方法,其中相應的二胺與光氣的反應在包含兩個區的反應器中進行,其中構成總反應器體積的大約20%至80%的第一區具有理想的混合,構成總反應器體積的80%至20%的第二區可以以活塞流為特徵。第二反應區優選為管式反應器的形式。
管式反應器用於氣相光氣化的優化是許多申請的主題,如在EP-A-0 570 799中使用噴射混合器原理公開了其原理(Chemie-Ing.-Techn. 44 (1972),第1055頁, 圖10)。
根據EP-A-1 362 847的教導,經管式反應器的環形空間供應的反應物料流的均化和兩種反應物料流在儘可能中心地進給到管式反應器中對反應區的穩定性和因此對整個氣相反應具有巨大的正面影響。
如EP-A-1 555 258中所述,所用管式反應器的擴大也使得必須擴大混合噴嘴,其通常為光滑噴射噴嘴的形式。但是,光滑噴射噴嘴的直徑擴大也由於所需的較大擴散長度而降低中心射流的混合速度並提高回混風險,這又導致形成聚合雜質和反應器上的固體附著物。根據EP-A-1 555 258的教導,當一個反應物料流經由另一反應物料流中同心安置的環形間隙高速噴射時,可以消除所述缺點。這能使用於混合的擴散長度小並使混合時間極短。該反應可以隨後以高選擇性進行以產生所需異氰酸酯。由此減輕聚合雜質的存在和附著物的形成。
根據EP-A-1 526 129的教導,中心噴嘴中的反應物料流的湍流增強對反應物的混合和因此對整個氣相反應具有正面影響。由於更好的充分混合,形成副產物的傾向降低。
EP-A-1 449 826公開了通過相應的二胺的光氣化製備二異氰酸酯的方法,其中將任選用惰性氣體或用惰性溶劑的蒸氣稀釋的氣態二胺和光氣分開加熱到200℃至600℃的溫度並在管式反應器中混合和反應,其中在管式反應器內布置平行於管式反應器的軸排列的數量n > 2的噴嘴,其中經n個噴嘴將包含二胺的料流供入管式反應器並經剩餘自由空間將光氣料流供入管式反應器。
管式反應器用於氣相光氣化時的進一步發展是WO 2007/028715的主題。所用反應器具有混合裝置和反應空間。根據WO 2007/028715的教導,該反應空間在前區包含混合空間,在此主要發生反應物氣態光氣和胺(任選與惰性介質混合)的混合,其通常伴隨著反應的開始。根據WO 2007/028715的教導,在反應空間的後部,隨後基本僅發生反應,並最多在微小程度上發生混合。優選地,在WO 2007/028715中公開的方法中,使用相對於流向旋轉對稱的反應空間,其可以在構造上基本分割成在流動方向上沿反應器縱軸的最多四個縱向區段(Längenabschnitt),其中這些縱向區段在流經的橫截面面積的尺寸上不同。
WO 2008/055898公開了通過相應的胺在反應器中在氣相中的光氣化製備異氰酸酯的方法,其中,類似於WO 2007/028715,所用反應器具有混合裝置和反應空間,該旋轉對稱的反應空間可以在構造上基本分割成在流動方向上沿反應器縱軸的最多四個縱向區段,其中這些縱向區段在流經的橫截面面積的尺寸上不同。但是,與WO 2007/028715相比,不藉助安裝到管式反應器中的體積物件(Volumenkörper)而是藉助反應器外壁的相應擴展或收縮實現流經的橫截面面積的變化。
EP-A-1 275 639同樣公開了,作為通過相應的胺與光氣在氣相中的光氣化製備異氰酸酯的可能的工藝變體,使用下述反應器:其中反應空間在流動方向上在這兩種反應物混合後具有流經的橫截面面積的擴展。通過橫截面面積的適當選擇的擴展,可以使反應混合物在反應器長度上的流速保持恰好恆定。這增加在相同反應器長度下可提供的反應時間。
EP-A-2 196 455公開了在包含相對於流動方向基本旋轉對稱的反應空間的反應器中在高於胺的沸點下使光氣和伯芳胺反應,其中在氨基轉化成異氰酸酯基團的轉化率為4%至80%的反應空間的區段中該反應混合物沿基本旋轉對稱的反應空間的軸的橫截面平均流速為最大8米/秒,且其中在反應空間的這一區段中該反應混合物沿基本旋轉對稱的反應空間的軸的橫截面平均流速始終低於在這一區段的起點處的橫截面平均流速。
EP-A-1 935 876公開了通過使相應的伯胺與光氣反應製備異氰酸酯的氣相法,其中在高於胺的沸點下在0.05至15秒的平均接觸時間內使光氣和伯胺反應,其中所述反應絕熱進行。
EP-A-2 408 738公開了如何可以避免光氣由於含光氣的料流在高溫下的過長停留時間而離解成氯氣和一氧化碳。通過將光氣在大於300℃的溫度下的停留時間減至最多5秒和通過將與光氣接觸的傳熱面的溫度限制為高於要建立的光氣溫度最多20 K,應避免這一點。
EP-B-1 935 875公開了通過使相應的伯胺與光氣在氣相中反應製備異氰酸酯的方法,其中通過將反應混合物經過噴入有液體的冷卻段導出反應空間,停止該反應,其中該冷卻段中的直接冷卻在串聯的兩個或更多個冷卻區中一步實現(所謂的反應混合物的「驟冷」)。
WO 2013/029918描述了通過使相應的胺與光氣反應製備異氰酸酯的方法,通過提高光氣與胺的比率或將一種或多種惰性物質添加到光氣和/或胺料流中,該方法也可毫無問題地在該氣相裝置的不同負荷下進行,特別是甚至在部分負荷範圍內運行該裝置時,該混合和/或反應也應在每種情況下優化的停留時間窗口內進行。此發明的方法應能以恆定的產品和工藝品質運行在不同負荷下的現有裝置。這應免去提供多個具有不同標稱容量的裝置。
該申請教導了為在標稱容量下運行生產裝置而優化光氣化的基本參數,特別例如反應物在各個裝置中的停留時間,這在低於標稱容量下運行該裝置時會造成產率和產品一致性方面的問題(參見第2頁第20至36行)。為了甚至在部分負荷(即與在標稱容量下的運行相比減小的胺料流)下也能達到優化的 - 窄 - 停留時間窗口,建議增加光氣料流和/或惰性成分(參見第3頁第5至19行),確切來說,優選使得所有組分的總流量基本相當於在標稱容量下的(參見第6頁第4至8行)。該申請在第2頁中提出的發明背景描述中儘管提到啟動程序,但完全沒有公開關於最有利地使非運行中的生產裝置(即胺料流和光氣料流等於0)達到標稱容量的所需運行狀態的具體操作的技術教導。該申請中公開的技術措施(即增加光氣料流和/或惰性成分)應僅就生產裝置在低於標稱容量下運行(即胺料流明顯大於0)的問題和如何有利地將在標稱容量下運行的裝置切換成在低於標稱容量下運行的問題進行考慮(參見實施例)。
儘管所述的現有技術方法成功地在不損失最終產物品質的情況下進行光氣化,但除了少數例外,所描述的方法僅是處於正常運行下的方法。沒有描述直到在待轉化的胺的所需流量下的穩定運行狀態前的啟動程序,即氣相光氣化裝置的啟動。
本領域技術人員知道由非運行中的生產裝置開始(例如在維護相關的停機後),連續運行的工業過程無法立即加轉回停產前的工藝參數。必須加熱反應物和裝置,可能必須將裝置惰性化,並將該裝置的反應物載量逐漸提高到所需目標值。氣相光氣化裝置的啟動(也稱為開動)是工業日常中經常發生的程序,其不一定與打開或另一機械介入光氣化裝置的方式相關聯。在實踐中,啟動的特徵在於,與在生產裝置的標稱容量下的連續運行相比,光氣相對於胺的過量可能偏差。特別在例如壓力波動造成回混時出現這樣的偏差。當要轉化的胺的當前流量與在該裝置的標稱容量下要轉化的胺的所需流量相比極小時,尤其觀察到這一點。光氣與胺的比率的這些量值波動不利,因為可沉澱出固體,如聚脲或胺鹽酸鹽。此外,在不當啟動的情況下,會不合意地形成胺微滴。氣相光氣化裝置的啟動因此是關鍵工藝步驟,因為此處的錯誤會嚴重幹擾實際連續生產(例如由於為了確保反應物和產物經過該裝置的足夠流量所需的壓力差的提高)。
因此,儘管在氣相光氣化領域中有各種進展,但仍需要進一步改進。考慮到這一需要,本發明提供一種運行用於使胺(2)與光氣(1)反應以產生相應的異氰酸酯(4)的氣相光氣化裝置(100)的方法,其中所述氣相光氣化裝置(100)至少包含
(i) 用於提供任選除光氣(1)外還包含惰性物質(3)的氣態光氣料流(10)的裝置1000,
(ii) 用於提供任選除胺(2)外還包含惰性物質(3)的氣態胺料流(20)的裝置2000,
(iii) 用於混合料流10和20的混合區(3100),其中所述混合區通過設備(1100、2100)分別與裝置1000和裝置2000相連,
(iv) 布置在混合區(3100)下遊的用於之前混合的料流10和20的進一步反應的反應區(3200),
(v) 布置在反應區(3200)下遊的用於終止所述反應的反應停止區(4000),
和任選的
(vi) 後處理部分(5000),其包含用於回收和再循環未轉化的光氣(1'')的設備(5100)和用於獲得以純淨形式製成的異氰酸酯的設備(5200),
其中,在氣相光氣化裝置(100)的正常運行中,光氣氣體料流(10)中的光氣(1)優選是新鮮光氣(1')和設備(5100)回收的再循環光氣(1'')的混合物,
其中通過運行下列步驟啟動氣相光氣化裝置(100):
(I) 在裝置1000中提供200℃至600℃,優選200℃至500℃,更優選250℃至500℃的溫度T10和100毫巴至3000毫巴,優選150毫巴至2800毫巴,更優選200毫巴至2500毫巴的絕對壓力p1的氣態光氣料流(10)並將這一氣態光氣料流(10)經設備1100連續引入混合區(3100)、反應區(3200)和反應停止區(4000),其中壓力p10大於混合區(3100)中的壓力p3100;
(II) 與步驟(I)同時或此後,
(a) 將溫度T3 < 200℃的惰性物質(3),優選在室溫和標準壓力下為液體的那些引入裝置2000,在裝置2000中加熱惰性物質(3)以獲得惰性氣體料流(30),使其經過設備2100、混合區(3100)、反應區(3200)和反應停止區(4000),或
(b) 將惰性氣體料流(30)
(b.1) 引入設備2100並從此處經過混合區(3100)、反應區(3200)和反應停止區(4000)
或優選
(b.2) 引入裝置2000並從此處經過設備2100、混合區(3100)、反應區(3200)和反應停止區(4000),
其中變體(a)和變體(b)中的惰性氣體料流(30)具有200℃至600℃,優選200℃至500℃,更優選250℃至500℃的溫度T30和100毫巴至3000毫巴,優選150毫巴至2800毫巴,更優選200毫巴至2500毫巴的絕對壓力p30,其中壓力p30在這兩種情況下都大於混合區(3100)中的壓力p3100;
(III) 在步驟(II)後,在裝置2000中提供200℃至600℃,優選200℃至500℃,更優選250℃至500℃的溫度T20和100毫巴至3000毫巴,優選150毫巴至2800毫巴,更優選200毫巴至2500毫巴的絕對壓力p20的氣態胺料流(20)並將這一氣態胺料流(20)經設備2100連續引入混合區(3100),其中壓力p20大於混合區(3100)中的壓力p3100,且其中料流(20)的組成和質量流量與料流(10)的組成和質量流量匹配,以使在混合區(3100)中,光氣(1)相對於胺(2)的伯氨基化學計算過量存在。
氣相光氣化根據本發明被理解為是指使胺光氣化成相應的異氰酸酯的一種工藝方案,其中胺在氣態下反應產生異氰酸酯並在該反應過程中,存在的所有組分(反應物、產物、中間產物、任選的副產物、任選的惰性物質)在經過反應區的過程中至少95.0質量%,優選至少98.0質量%,更優選至少99.0質量%,再更優選至少99.8質量%,特別是至少99.9質量%保持在氣相中,在每種情況下基於存在的所有組分的總質量計。
合適的胺(2)尤其是異佛爾酮二胺、六亞甲基二胺、雙(對氨基環己基)甲烷、甲苯二胺和二苯基甲烷二胺。
在本發明中,術語「氣相光氣化裝置的啟動」包括使非運行的氣相光氣化裝置(例如在維護相關的停機後)達到以要轉化的胺的所需流量M'目標(2) [例如t(胺)/h]表示的所需生產量所必需的所有工藝步驟。氣相生產裝置(100)在M'目標(2)下的運行在本發明中被稱作正常運行。M'目標(2)可以,但不需要,相當於在氣相生產裝置(100)的標稱容量M'標稱(2)下的M'目標(2)的值。生產裝置的標稱容量在專業領域中以每年要生產的產品噸數(「每年噸數」)表示,其中考慮所有可計劃的裝置停機。
本發明中關於可數參數的詞語「一」僅被理解為不定冠詞並且只有在明確說明時才是數詞,例如通過加上「剛好一個」。例如,短語「一反應區」不排除存在多個(串聯或並聯)反應區的可能性。
對本發明而言重要的是,通過首先向其中充填光氣而啟動光氣化裝置(100)。在啟動過程中,始終在光氣後加入胺,這防止胺回混到光氣路徑中並確保光氣相對於胺的過量。在胺轉化成氣相之前和優選在此過程中,另外引入惰性物質,以防止在胺供應開始時光氣回流到胺進料中(回混)。
下面詳細闡述本發明的步驟。各種實施方案在此可以任意互相組合,除非本領域技術人員從上下文中看出相反的意思。
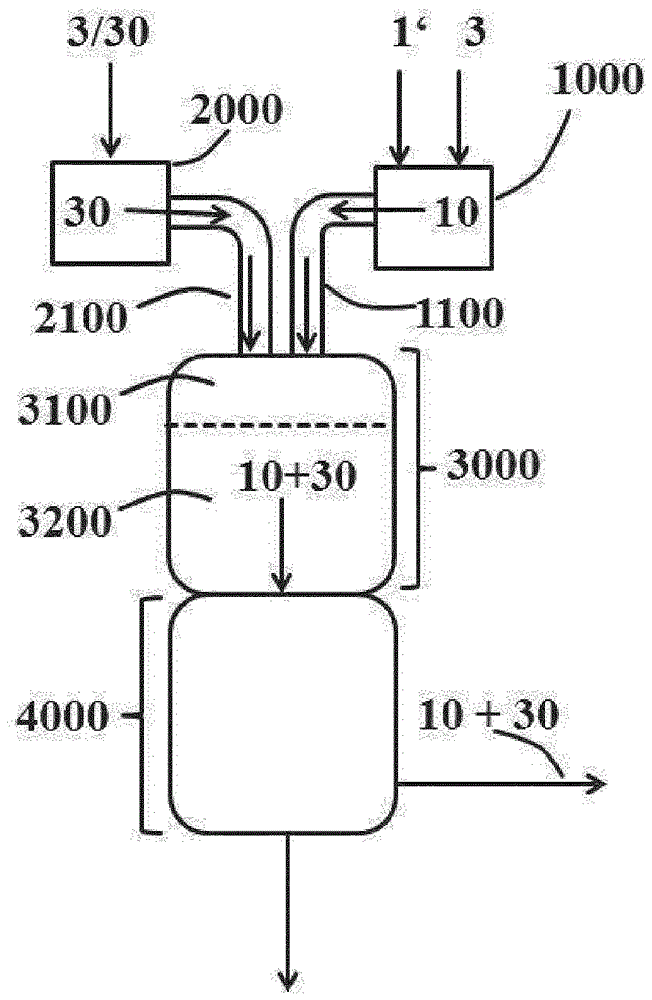
根據本發明,氣相光氣化裝置(100)至少包含上文在(i)至(v)下列舉的設備(也參見圖1,其顯示要根據本發明使用的氣相光氣化裝置(100)的設備,包括優選存在的後處理部分和在正常運行中的物質料流)。
(i) 作為用於提供氣態光氣料流的裝置(1000),原則上可以使用現有技術中已知並適用於將光氣轉化成氣相的各種裝置。優選通過如DE 10 2009 032413 A1段落[0081]至[0118]中所述在蒸餾塔中的蒸餾或部分蒸發生成光氣氣體。可以由各種可想到的蒸發器,例如自然循環蒸發器、升膜蒸發器和降膜蒸發器實現在塔底的能量供應。降膜蒸發器尤其優選。
(ii) 作為用於提供氣態胺料流的裝置(2000),原則上可以使用現有技術中已知並適用於將胺轉化成氣相的各種裝置,如本領域技術人員已知的蒸發裝置。在一個優選實施方案中,裝置2000包含蒸發設備和用於隨後使胺(2)過熱的設備。非常特別優選的是多級蒸發和過熱系統,其中在蒸發和過熱系統之間安裝微滴分離器和/或該蒸發裝置也具有微滴分離器的功能。合適的微滴分離器描述在例如"Droplet Separation", A. Bürkholz, VCH Verlagsgesellschaft, Weinheim – New York – Basle – Cambridge, 1989中。在離開流動方向上的最後一個過熱器後,將預熱至其目標溫度的氣態反應物料流(20)供入反應空間。
(iii) 可根據本發明使用的混合區(3100)可以以本領域技術人員已知的方式,優選如EP-A-2 196 455,尤其是段落[0047]至[0049]中,和EP-A-1 935 876,尤其是段落[0027]至[0029]中所述那樣構造。混合區在正常運行中料流(10)和(20)初次彼此會合之處開始。
(iv) 在混合區(3100)中初次彼此會合的胺和光氣氣體料流在停留時間裝置(Verweilzeitapparat),即反應區(3200)中進一步反應。混合區(3100)和反應區(3200)也可優選如EP 2 196 455 A1,尤其是段落[0042]至[0049]中所述那樣合併在單一裝置,即反應器(3000)中。
根據本發明,將用於提供氣態光氣氣體料流(1000)和胺氣體料流(2000)的裝置與混合區(3100)的設備1100和2100是適用於將來自裝置1000和2000的氣體料流(10)和(20)轉移到混合區(3100)中的設備。這些設備除用於輸送氣體料流的管線外還優選包含確保光氣氣體料流(10)和胺氣體料流(20)在混合區(3100)中的強力充分混合的噴嘴裝置。可以將氣體料流(10)和(20)的每個各自噴入混合區(3100)。但是,優選的是設備1100和2100的管線通往共用的噴嘴裝置的實施方案(未顯示在圖1中)。在這一實施方案中,這兩個氣體料流之一,優選胺氣體料流(20)經由布置在優選圓柱形容器的中心的內部噴嘴供入混合區(3100)。另一氣體料流,優選光氣氣體料流(10)經過由內部噴嘴的外壁與該容器的內壁形成的環形空隙引入。這兩個氣體料流在內部噴嘴的出口孔處(= 混合區的起點)混合。這一實施方案例如顯示在EP-A-1 449 826的圖1和EP-A-1 362 847的圖1中。在這種情況下,設備1100和2100部分互相集成併集成到混合區(3100)中。也可以如EP-A-1 449 826的圖2中所示,使用由幾個獨立噴嘴構成的裝置代替單一的中心噴嘴。例如在EP-A-2 196 455,尤其是段落[0047]至[0048]中,和EP-A-1 935 876,尤其是段落[0027]和[0028]中描述了可根據本發明用於設備1100和2100的進一步實施方案。
(v) 可根據本發明使用的反應停止區(4000)是本領域技術人員已知的。優選的是如EP 1 935 875 B1,尤其是段落[0024]和[0025]中所述的實施方案。優選地,該反應停止區最晚在料流20在步驟(III)中首次進入混合區(3100)時投入運行。在反應停止區(4000)中,將除異氰酸酯(4)外基本還包含氯化氫副產物(Koppelprodukt)和未轉化的光氣的反應粗產物(40)迅速冷卻,優選通過將惰性溶劑(優選鄰二氯苯,ODB)任選與一部分之前形成並再循環的異氰酸酯(4)一起噴入氣體料流(40)。優選地,粗反應產物(40)在反應停止區(4000)中分離成氣態組分(蒸氣,50)和液體組分(60)。
在本發明的方法的一個特別優選的實施方案中,在反應停止區(4000)中獲得的粗產物在相同的氣相光氣化裝置(100)中後處理以從液體混合物(60)中分離異氰酸酯(4)。在這種情況下,氣相光氣化裝置(100)另外包含
(vi)後處理部分(5000) 。
適用於後處理的裝置描述在WO 2011/003532,尤其是第5頁第19行至第28頁第5行,以及EP 1 371 636 B1、EP 1 371 635 B1和EP 1 413 571 B1中,在每種情況下為全文。後處理部分(5000)可分成用於回收和再循環未轉化的光氣(和用於分離氯化氫副產物)的設備(5100)和用於獲得以純淨形式製成的異氰酸酯(和任選用於再循環惰性溶劑)的設備(5200)。在圖1中僅示意性標示後處理部分而沒有下文給出的細節。特別地,後處理部分(5000)包含用於通過用惰性溶劑洗滌而從來自反應停止區(4000)的蒸氣(50)中除去異氰酸酯的洗滌塔(5110)、用於通過吸收在惰性溶劑中(這導致氯化氫和惰性物(70)與光氣分離)而從洗滌塔(5110)的蒸氣中回收光氣的光氣吸收塔(5120)、用於分離光氣和惰性溶劑的光氣解吸塔(5130)、尤其用於從粗製異氰酸酯中分離低沸物(尤其是來自反應停止區的惰性溶劑)的溶劑塔(5210)、尤其用於從在溶劑塔中預提純的異氰酸酯中分離高沸物(例如含聚脲的殘留物)以獲得純化的最終產物的精細提純塔(5220)。
可以(未顯示在圖1中)將用於提供氣態光氣料流(10)的裝置(1000)集成到光氣解吸塔(5130)中以使來自後處理的含溶劑的光氣料流與新鮮光氣(1')一起蒸發並在光氣解吸塔(5130)中蒸餾。所得氣態光氣經設備1100供入混合區,同時優選將分離出的惰性溶劑導入洗滌塔(5110)和/或光氣吸收塔(5120)。
在本發明的方法的步驟(I)中(也參見圖2),在裝置1000中,提供氣態光氣料流(10)並經設備1100連續引入混合區(3100)、反應區(3200)和反應停止區(4000),其中壓力p10大於混合區(3100)中的壓力p3100。在此過程中,中斷胺氣體料流(20)的供應。優選地,在步驟(I)的進行過程中,裝置部分——設備1100、混合區(3100)、反應區(3200)和反應停止區(4000)不含雜質,尤其不含在之前的生產周期中可能來自反應停止的已由於再循環光氣料流而分布在光氣化裝置中或有意作為惰性物質添加的溶劑。優選地,在步驟(I)中就將光氣氣體質量流量M'(10)調節到在所需容量下的連續生產過程中的稍後目標值M'目標(10)。氣態光氣料流(10)除光氣(1)外還可含有惰性物質(3)。在裝置1000中,在200毫巴至3000毫巴的絕對壓力p30下將光氣氣體料流(10)加熱到200℃至600℃的溫度T30。T10和p10的值涉及整個料流(10),即任選地涉及離開設備1100時的光氣(1)和惰性物質(3)的混合物。可根據本發明使用的惰性物質(3)除在室溫和標準壓力下已為氣態的物質,如氮氣、氦氣或氬氣外還包含在室溫和標準壓力下為液體的惰性有機溶劑的蒸氣,例如任選具有滷素取代的芳烴,例如氯苯或二氯苯(所有異構體,優選鄰二氯苯)。特別優選使用氮氣稀釋光氣。可以以現有技術中常規的方式選擇惰性物質(3)在光氣氣體料流(10)中的含量。在步驟(I)中,在光氣循環下加熱光氣化裝置(100),接著在步驟(II)中在胺側上形成惰性氣體料流。
在步驟(I)中,光氣料流(10)中的光氣優選至少部分來自氣相光氣化裝置的後處理部分(5000)(來自之前的生產周期的再循環光氣(1''))。優選如WO 2011/003532,尤其是第5頁第19行至第28頁第5行中所述進行後處理中的光氣回收。
在本發明方法的步驟(II)中(也參見圖3),將惰性氣體料流(30)至少導過設備2100(並從此處經過混合區(3100)、反應區(3200)和反應停止區(4000))。優選地,以將惰性氣體料流(30)從裝置2000導入設備2100(並從此處經過混合區(3100)、反應區(3200)和反應停止區(4000))的方式將裝置2000和設備2100惰性化。在此步驟的過程中打開任選存在於裝置2000和設備2100之間(以及設備2100和混合區3100之間)的阻隔設備,以使惰性氣體料流(30)可從蒸發裝置2000流出到設備2100中(並從此處流入混合區(3100))。
可通過(a)將溫度T3 99.97%的純度(氣相色譜法,GC)、< 5 ppm的ODB殘留溶劑含量(GC)、< 10 ppm的可水解氯的殘留氯含量(根據ASTM D4663滴定)、< 5 ppm的結合氯的酸度(根據ASTM D5629滴定)和作為Hazen值測得的< 15的色值(根據DIN EN ISO 6271測定)。
對比例1: 在光氣(1)前供入胺(2)以啟動氣相光氣化裝置(100)
氣相光氣化裝置(100)如上所述運行。在停機後,如下重啟該裝置:胺蒸發器(2000)和包括胺噴嘴的導管(2100)在380℃的設定溫度下充填氮氣氣體料流(30)。光氣化反應器(3000)不含反應物和產物並用熱氮氣(30)惰性化。啟動胺蒸發器(2000)中的胺蒸發,並使TDA在300℃下蒸發,在進一步熱交換器中加熱至410℃並在1683毫巴的絕對壓力下作為氣態TDA (20)經胺噴嘴噴入光氣化反應器(3000)。在45分鐘的計劃啟動期過程中引入光氣化反應器(3000)的TDA (2)的量應從0 t/h連續提高到12 t/h。在開始供應胺後5分鐘,將光氣氣體料流(10)在61 t/h的質量流量、在反應器入口處320℃的溫度和1683毫巴的絕對壓力下噴入光氣化反應器(3000)。在20分鐘後,由於在反應物TDA氣體料流(20)和光氣氣體料流(10)進入光氣化反應器(3000)的入口和底部(4100)的蒸氣出口之間的壓差迅速升高到大於1000毫巴而非在正常運行中的10毫巴,必須停止該裝置,且蒸發光氣(1)和TDA (2)所需的蒸發能量不能再提高。通過關閉光氣和TDA供應而中斷該啟動。在打開光氣化裝置後,發現在底部(4100)被TDA和聚脲堵塞的除霧器和在驟冷器(Quenchen)(4000)中被沉積物大面積覆蓋的表面。
對比例2: 同時供入胺(2)和光氣(1)以啟動氣相光氣化裝置(100)
氣相光氣化裝置(100)如上所述運行。在停機後,如下重啟該裝置:胺蒸發器(2000)和包括胺噴嘴的導管(2100)在380℃的設定溫度下充填氮氣氣體料流(30)。光氣化反應器(3000)不含反應物和產物並用熱氮氣(30)惰性化。啟動胺蒸發器(2000)中的胺蒸發,並使TDA在300℃下蒸發,在進一步熱交換器中加熱至410℃並在1683毫巴的絕對壓力下作為氣態TDA (20)經胺噴嘴噴入光氣化反應器(3000)。在45分鐘的計劃啟動期過程中引入光氣化反應器(3000)的TDA (2)的量應從0 t/h連續提高到12 t/h。與TDA氣體料流(20)經胺噴嘴送入光氣化反應器(3000)同時,打開光氣供應並將光氣氣體料流(10)在61 t/h的質量流量、在反應器入口處320℃的溫度和1691毫巴的絕對壓力下噴入光氣化反應器(3000)。該裝置在45分鐘啟動時間後可以以正常運行模式運行。在5天後,由於在反應物TDA氣體料流(20)和光氣氣體料流(10)進入光氣化反應器(3000)的入口和底部(4100)的蒸氣出口之間的壓差升高到793毫巴而非在正常運行中的10毫巴,必須停止該裝置,且蒸發光氣(1)和TDA (2)所需的蒸發能量幾乎無法提高。在該裝置停止和打開後,在胺噴嘴口處、沿反應器空間的表面和在驟冷器表面上發現含聚脲的嚴重(massiv)沉積物。
對比例3: 在胺(2)前供入光氣(1)以啟動氣相光氣化裝置(100),但不用惰性氣體料流(30)將胺供應設備惰性化
通過使來自出自反應停止區(4000)的蒸氣(50)在320℃下的後處理(5100)的再循環光氣(1'')經過光氣化反應器(3000)、反應停止區(4000)回到該後處理,建立光氣的循環。在反應停止區(4000)中,在此過程中,只有在反應混合物(40)的流動方向上的第二驟冷在運行,由此將光氣料流冷卻。在這一光氣循環中,使61 t/h的光氣循環(步驟(I))。在此過程中,不用氮氣流(30)衝洗胺蒸發器(2000)和包括胺噴嘴的導管2100。光氣化反應器(3000)一旦調溫到320℃,就通過將預熱到220℃的液體TDA (2)與氮氣一起送入胺蒸發器(2000)、在其中藉助熱交換器在300℃下蒸發並然後藉助另一熱交換器將其加熱到410℃而開始胺蒸發 – 不預先用氮氣氣體料流(30)衝洗包括胺噴嘴的導管(2100)。由此獲得的TDA料流(20)在1654毫巴的絕對壓力下經胺噴嘴噴入光氣化反應器(3000)(步驟(III))。在氣相光氣化裝置的啟動過程中(即直到胺質量流量達到M'目標(2),這在45分鐘後發生)引入光氣化反應器(3000)的TDA (2)的量從0 t/h連續增加到12 t/h,其中在啟動結束時光氣化反應器(3000)中的工作壓力為1641毫巴(絕對)。在TDA氣體料流(20)在啟動過程中朝光氣化反應器(3000)的方向初次離開胺噴嘴前不久,使在反應混合物(40)的流動方向上的第一驟冷投入運行。通過新鮮光氣(1')和在後處理中回收的光氣(1'')的混合物替代在該裝置啟動後消耗的光氣。在60分鐘後,15.6 t/h的TDI (4)離開後處理階段的最後一個蒸餾塔。
在啟動後,胺蒸發(2000)和光氣化反應器(3000)之間的壓差始終進一步提高。在3小時後,由於胺噴嘴處的工作壓力已升至2.5巴(絕對),必須停止光氣化反應器(3000)。在該裝置停止和打開後,在胺噴嘴的出口孔中和在通往胺噴嘴的管線中發現炭化的殘留物。這可歸因於在啟動階段中光氣回流到設備2100中,以致形成堵塞胺噴嘴的出口孔的TDI沉積物。
實施例4(本發明)
如實施例3中所述操作,只是一旦將光氣化反應器(3000)調溫到320℃,就在380℃的設定溫度下用熱氮氣(30)衝洗胺蒸發器(2000)和包括胺噴嘴的導管(2100)(步驟(II))。在60分鐘後,15.6 t/h的TDI (4)離開後處理(5200)的最後一個蒸餾塔。
在這一程序中,防止在啟動期過程中的胺噴嘴的堵塞和在光氣化反應器(3000)中形成沉積物,以使裝置(100)可長時間運行多達超過一年。明顯減少不想要的副產物,如聚脲等的形成並且可以不發生該啟動產品與更高純度的TDI的稍後摻混。