一種基於菲並‑S,S‑二氧‑二苯並噻吩單元的共軛聚合物及其製備方法與應用與流程
2024-02-29 18:04:15 2
本發明屬於有機光電材料領域,具體涉及一種基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物及其製備方法與應用。
背景技術:
在過去的三十年中,有機電子和光電子產業,包括有機/聚合物發光二極體(pled),有機場效應電晶體,有機太陽能電池等領域得到了迅猛的發展,並逐漸實現產業化。有機電子產品具有價格低廉,體輕便攜等優點。使其具有極大的市場潛力。因此開發具有市場吸引力的有機電子產品吸引了世界上眾多研究機構和科研團隊的關注,而在這其中,開發新型高效穩定的材料成為關鍵。
但是,目前有機發光器件技術在發展過程中遇到了瓶頸問題,就是發光器件的發光效率和使用壽命達不到實用化要求,這大大限制了oled技術的發展,針對這一個問題,各個研究機構都在進行探索性的研究。
本發明所涉及到的菲並-s,s-二氧-二苯並噻吩單元及其聚合物,因為具有較好的溶解性能,適用於溶液加工,以及較好的螢光量子產率,其發光器件不僅高效穩定,而且為更藍的飽和藍光,可以同時實現發光器件的發光效率和使用壽命的提高,可以滿足全彩顯示的要求。所以在有機電子顯示領域有巨大的發展潛力和前景。
技術實現要素:
本發明的目的在於針對目前聚合物發光二極體面臨的問題,提供一種基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物。該共軛聚合物可用作發光材料,且具有較好的溶解性,較高的螢光量子產率,適合於溶液加工和噴墨列印,具有良好的發展前景。
本發明的目的還在於提供所述一種基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物的製備方法。
本發明的目的還在於提供所述一種基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物在製備發光二極體的發光層中的應用。
一種基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物,具有如下化學結構式:
式中,r1-r4為氫、芳基、三苯胺、碳原子數1-20的直鏈或支鏈烷基,或為碳原子數1-20的烷氧基;0≤x≤1,聚合度n為1-300;
ar為如下結構中的任意一種:
2,7-取代芴;
3,6-取代芴;
2,7-取代矽芴;
3,6-取代矽芴;
2,7-取代螺芴;
3,6-取代螺芴;
2,7-取代-9,9-二烷氧基苯基芴;
3,6-取代-9,9-二烷氧基苯基芴;
2,7-取代咔唑;
3,6-取代咔唑;
2,6-取代-二噻吩並噻咯;
2,6-取代-二噻吩並環戊二烯;
2,5-取代吡啶;
2,6-取代吡啶;
3,5-取代吡啶;
3,5-雙(4-取代-苯基)-4-基-1,2,4-三唑;
3,5-雙(4-取代-苯基)-1,2,4-噁二唑;
4,7-雙(5-取代-4-烷基噻吩基)-2,1,3-苯並噻二唑;
4,7-雙(5-取代-4-烷基噻吩基)2,1,3-苯並硒二唑;
4,7-取代-5,6-烷基-2,1,3-苯並噻二唑;
4,7-取代-5,6-烷基-2,1,3-苯並硒二唑;
2,5-取代-3,4-二烷基噻吩;
2,5-取代-3,4-二烷基硒吩;
5,5-取代-4,4-二烷基-聯噻吩;
茚芴;
吲哚咔唑;
4,9-取代-6,7-烷基-菲並噻二唑;
4,9-取代-6,7-烷基-菲並硒二唑;
萘並茚芴;
其中,r為h、芳基、三苯胺、碳原子數1-20的直鏈或者支鏈烷基,或為碳原子數1-20的烷氧基。
製備所述的一種基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物的方法,包括如下步驟:
將菲並-s,s-二氧-二苯並噻吩單體與含ar結構的硼酸酯單體通過suzuki聚合反應後,再依次採用苯硼酸和溴苯進行封端反應,得到所述基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物。
進一步地,所述菲並-s,s-二氧-二苯並噻吩單體與含ar結構的硼酸酯單體的摩爾比為1:1~1:5。
進一步地,所述suzuki聚合反應的溫度為80~100℃,時間為24~48小時。
進一步地,採用苯硼酸和溴苯進行封端反應的溫度均為80~100℃,時間均為12~24小時。
所述的一種基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物應用於製備發光二極體的發光層,將基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物用有機溶劑溶解,再通過旋塗、噴墨列印或印刷成膜,得到所述發光二極體的發光層;基於該發光層的發光二極體可用於製備平板顯示器。
進一步地,所述有機溶劑包括氯苯。
與現有技術相比,本發明具有以下優點:
(1)本發明的基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物,由於具有較大的共軛長度,所以有較高的螢光量子產率,有利於提高材料的器件效率;
(2)本發明的基於菲並-s,s-二氧-二苯並噻吩單元的共軛聚合物,具有較好的溶解性,基於該聚合物的發光層在製備電致發光器件時不用退火處理,使得製備工藝更簡單。
附圖說明
圖1為聚合物p1的熱分析譜圖;
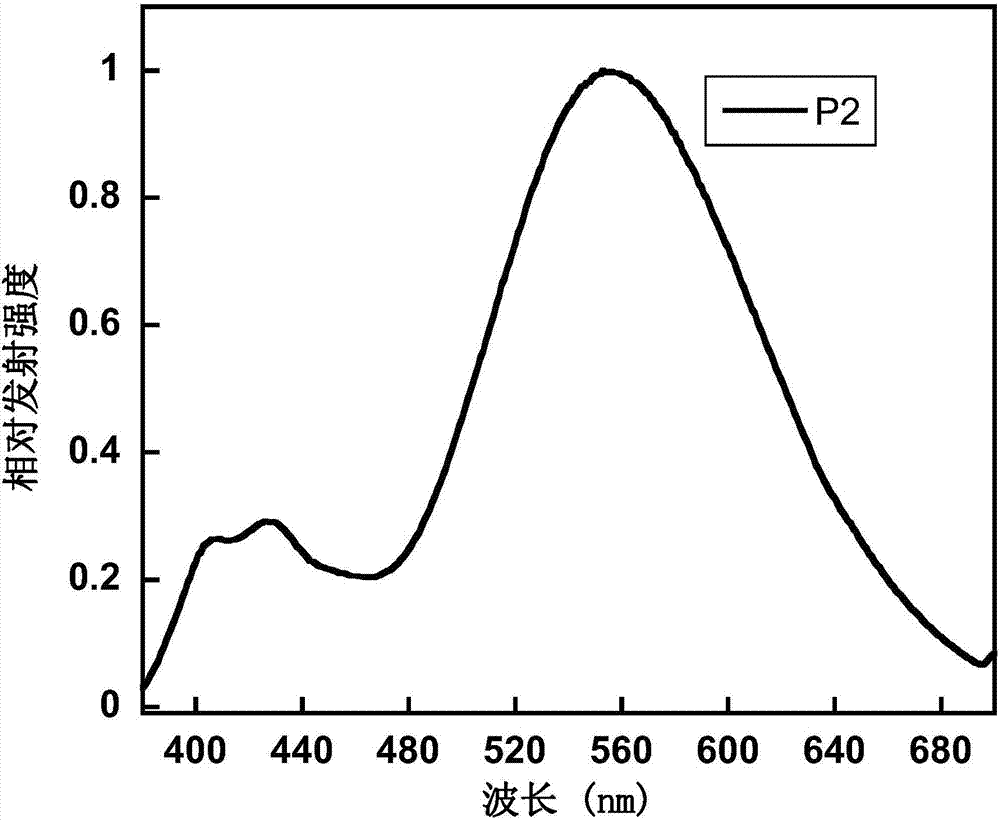
圖2為聚合物p2在薄膜狀態下的光致發光光譜圖;
圖3為基於聚合物p3的電致發光器件的電流密度-流明效率譜圖。
具體實施方式
下面結合實施例,對本發明作進一步地詳細說明,但本發明的實施方式不限於此。
實施例1
1-溴二菲甲酸甲酯的製備
在氬氣氣氛下,將1-溴-2-菲甲酸(10g,37.83mmol)加入兩口瓶中,再加入100ml甲醇,然後逐滴加入濃硫酸(39.06mg,378.29umol),加熱到110℃,反應18h;將反應混合物倒入水中,用乙酸乙酯萃取,有機層用食鹽水完全洗滌後,加無水硫酸鎂乾燥。溶液濃縮後,得到白色固體粗品,用矽膠柱層析提純(洗脫劑選擇石油醚/二氯甲烷=3/1,v/v),產物放置冰箱中,得到白色固體,產率85%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物,製備過程化學反應方程式如下所示:
實施例2
2-溴硫芴的製備
在氬氣氛圍下,將硫芴(20g,108.54mmol)加入到250ml兩口瓶中,再加入100ml氯仿進行完全溶解,加入0.5g(275mg,1.09)碘單質,在避光的情況下,逐滴加入液溴(38.16g,238.80mmol),反應液在0℃冰浴下攪拌2小時,然後在室溫下攪拌2小時,加入飽和的亞硫酸氫鈉淬滅液溴,將反應混合物倒入水中,用乙酸乙酯萃取,有機層用食鹽水完全洗滌後,加無水硫酸鎂乾燥。溶液濃縮後,得到白色固體粗品,然後用氯仿重結晶,產率85%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物,製備過程化學反應方程式如下所示:
實施例3
2-硼酸酯硫芴的製備
在氬氣氣氛下,將2-溴硫芴(10g,29.24mmol)溶解於180ml精製的四氫呋喃(thf)中,在-78℃下逐漸滴加1.6moll-1的正丁基鋰28ml,反應2小時,然後快速加入2-異丙氧基-4,4,5,5-四甲基-1,3,2-二氧雜硼烷25ml,在-78℃下繼續反應1小時,緩慢升溫至室溫反應24小時。將反應混合物倒入水中,用乙酸乙酯萃取,有機層用食鹽水完全洗滌後,加無水硫酸鎂乾燥。溶液濃縮後,得到淺黃色粘稠狀粗品,用矽膠柱層析提純(洗脫劑選擇石油醚/乙酸乙酯=20/1,v/v),產物放置箱中,得到白色固體,產率70%。1hnmr和gc-mass測試表明為目標產物,製備過程化學反應方程式如下所示:
實施例4
化合物m1的製備
在氬氣氛圍下,將2-硼酸酯硫芴(5g,10.46mmol)和1-溴-2-菲甲酸甲酯(7.6g,28.66mmol)加入到兩口瓶中,再加入100ml甲苯進行完全溶解,再加入碳酸鈉(6.07g,57.32mmol)、四丁基溴化銨(312.01mg,967.86umol)和四三苯基磷鈀(264.93mg,229.26umol),在110℃下反應18h。將反應混合物倒入水中,用乙酸乙酯萃取,有機層用食鹽水完全洗滌後,加無水硫酸鎂乾燥。溶液濃縮後,用矽膠柱層析提純(洗脫劑選擇石油醚/二氯甲烷=7/1,v/v),最終得到白色固體,產率80%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物m1,製備過程化學反應方程式如下所示:
實施例5
化合物m2的製備
在氬氣氛圍下,將化合物m1(10g,23.89mmol)加入單口瓶中,再加入50ml無水thf直到完全溶解;再將反應液在0℃下反應1h,再逐滴加入正辛基溴化鎂(25.98g,119.47mol,c8h17mgbr),混合液在室溫下反應18h。將水加入到反應液中來淬滅反應,用乙酸乙酯萃取,有機層用食鹽水完全洗滌後,加無水硫酸鎂乾燥;溶液濃縮後,用矽膠柱層析提純(洗脫劑選擇石油醚/二氯甲烷=3/1,v/v),產物放置冰箱中,得到白色固體,產率80%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物m2,製備過程化學反應方程式如下所示:
實施例6
化合物m3的製備
在氬氣氛圍下,將化合物m2(5g,4.09mmol)溶於50ml二氯甲烷中,在室溫下逐滴加入三氟化硼乙醚溶液(439.59mg,6.48mmol),反應18h;用乙酸乙酯萃取,有機層用食鹽水完全洗滌後,加無水硫酸鎂乾燥;溶液濃縮後,用矽膠柱層析提純(洗脫劑選擇石油醚),產物放置冰箱中,得到白色固體,產率90%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物m3,製備過程化學反應方程式如下所示:
實施例7
化合物m4的製備
在氬氣氛圍下,將化合物m3(5g,4.50mmol)溶於50ml二氯甲烷中,再加入鐵粉(185.35mg,3.32mmol),再逐滴加入液溴(1.93g,12.10mmol),在室溫下反應18h;用乙酸乙酯萃取,有機層用食鹽水完全洗滌後,加無水硫酸鎂乾燥;溶液濃縮後,用矽膠柱層析提純(洗脫劑選擇石油醚),產率70%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物m4,製備過程化學反應方程式如下所示:
實施例8
化合物m5的製備
在氬氣氛圍下,將化合物m4(10g,13.25mmol)加入到250ml兩口瓶中,加入100ml乙酸進行溶解,再加入雙氧水(1.35g,39.75mmol,h2o2),加熱到80℃,反應16小時;用乙酸乙酯萃取,有機層用食鹽水完全洗滌後,加無水硫酸鎂乾燥;溶液濃縮後,用矽膠柱層析提純(洗脫劑選擇石油醚),產率75%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物m5,製備過程化學反應方程式如下所示:
實施例9
化合物m6的製備
在氬氣氣氛下,將化合物m5(10g,18.24mmol)溶解於180ml精製的thf中,在-78℃下逐漸滴加1.6mol/l的正丁基鋰28ml,反應2小時,然後快速加入2-異丙氧基-4,4,5,5-四甲基-1,3,2-二氧雜硼烷25ml,在-78℃下繼續反應1小時,緩慢升溫至室溫反應24小時;將反應混合物倒入水中,用乙酸乙酯萃取,有機層用食鹽水完全洗滌後,加無水硫酸鎂乾燥;溶液濃縮後,得到淺黃色粘稠狀粗品,用矽膠柱層析提純(洗脫劑選擇石油醚/乙酸乙酯=20/1,v/v),產物放置冰箱中,得到白色固體,產率70%。1hnmr和gc-mass測試表明為目標產物m6,製備過程化學反應方程式如下所示:
實施例10
2,7-二溴芴的製備
在250ml三口瓶中,加入芴(24.5g,0.1mol)、鐵粉(88mg,1.57mmol)、三氯甲烷100ml;冰水浴冷卻,滴加溴(17.6g,0.1mol)/三氯甲烷混合溶液35ml;滴加時瓶內溫度不超過5℃;反應完畢,過濾、氯仿重結晶,得白色固體20.3g,產率83%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物,製備過程化學反應方程式如下所示:
實施例11
2,7-二溴-9,9-二辛基芴的製備
在三口瓶中加入2,7-二溴芴(9.7g,0.03mol)、苄基三乙基氯化銨(0.07g,0.3mmol)、二甲基亞碸90ml和45ml氫氧化鈉水溶液(50wt%),室溫下攪拌形成懸浮液;加入1-溴正辛烷(12.5g,65mmol),繼續攪拌3小時後,用乙醚萃取;用飽和氯化鈉水溶液洗滌乙醚相,無水硫酸鎂乾燥;蒸去溶劑,產物用石油醚作洗脫劑柱層析提純,得白色固體。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物,製備過程化學反應方程式如下所示:
實施例12
2,7-二硼酸酯-9,9-二辛基芴的製備
在氬氣氣氛下,將2,7-二溴-9,9-二辛基芴(5g,9.12mmol)溶解於180ml精製的thf中,在-78℃下逐漸滴加1.6mol.l-1的正丁基鋰28ml,反應2小時,然後加入2-異丙氧基-4,4,5,5-四甲基-1,3,2-二氧雜硼烷25ml,在-78℃下繼續反應1小時,然後升溫至室溫反應24小時;將反應混合物倒入水中,用乙酸乙酯萃取,有機層用食鹽水完全洗滌後,加無水硫酸鎂乾燥;溶液濃縮後,得到淺黃色粘稠狀粗品,用矽膠柱層析提純(洗脫劑選擇石油醚/乙酸乙酯=15/1,v/v),產物放置冰箱中,得到白色固體,產率70%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物,製備過程化學反應方程式如下所示:
實施例13
3,6-二溴咔唑的製備
在500ml兩口瓶中加入咔唑(24.7g,0.1mol),二甲基甲醯胺200ml,攪拌至完全溶解,n-溴代琥珀醯亞胺(nbs,49.84g,0.28mol)用120mln,n-二甲基甲醯胺溶解,冰浴至0℃,滴加nbs溶液,反應,避光,滴加完畢後,讓溫度自動上升至室溫後,反應6小時,將反應液滴加到水中沉澱,抽濾得到粗產物後,將抽濾物用無水乙醇進行重結晶,烘乾,得到寶色針狀固體,產率85%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物,製備過程化學反應方程式如下所示:
實施例14
3,6-二溴-n-辛基咔唑的製備
在250ml三口瓶中加入3,6-二溴咔唑(16.25g,0.05mmol)、甲苯100ml、四丁基溴化銨(0.8g,3.5mmol),攪拌溶解,然後滴加50wt%koh水溶液11ml,然後再加入溴辛烷(19.3g,0.1mol),在80℃下反應24小時,加水終止反應,水洗分離出來的有機相,水相用二氯甲烷萃取後,合併有機相,用無水mgso4乾燥,減壓蒸餾除去溶劑後得到淺黃色固體,用石油醚重結晶得到白色粉末固體。產率90%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物,製備過程化學反應方程式如下所示:
實施例15
3,6-二(4,4,5,5-四甲基-1,3,2-二氧雜硼烷-二基)-n-辛基咔唑的製備
在三口瓶中加入3,6-二溴-n-辛基咔唑(13.11g,30mmol)和新蒸的乙醚250ml,攪拌完全溶解至澄清透明後,將反應液冷卻至-78℃,然後一次性加入2-異丙氧基-(4,4,5,5-四甲基)-1,3,2-乙二氧基硼酸酯(37ml,180mmol),在-78℃下攪拌2小時,再將溫度升至室溫,反應24小時後結束反應;用乙醚萃取,飽和食鹽水洗滌4次,再用無水硫酸鎂乾燥,過濾後,蒸餾除去溶劑,產物用石油醚/乙酸乙酯(10:1,v/v)為洗脫劑柱層析提純,得到白色固體,產率45%。1hnmr、13cnmr、ms和元素分析結果表明所得到的化合物為目標產物,製備過程化學反應方程式如下所示:
實施例16
聚合物p1的製備
在氬氣氛圍下,將2,7-二硼酸酯-9,9-二辛基芴(300mg,272.92μmol)和菲並-s,s-二氧-而苯並噻吩(342.92mg,272.92μmol)加入100ml兩口瓶內,再加入8ml甲苯進行完全溶解,再加入醋酸鈀(2.45mg,10.92μmol)和三環己基膦(6.12mg,21.83μmol),然後加入2ml四乙基氫氧化銨,升溫至80℃,反應24小時;然後加入30mg苯硼酸進行封端,12小時後,再用0.1ml溴苯進行封端;繼續反應12小時之後,將產物滴加在甲醇中沉澱出來,攪拌,過濾,再將粗產物溶於20ml的甲苯中,以200~300目矽膠為固定相,用甲苯為洗脫劑進行柱層析,再將溶劑減壓濃縮後,再一次在甲醇中沉析出來,攪拌,過濾,真空乾燥後得到聚合物固體;最後再依次用甲醇、丙酮、四氫呋喃各抽提24小時,除去小分子;將濃縮後的四氫呋喃溶液滴入甲醇中沉析,真空乾燥後得到的纖維狀固體共軛聚合物p1。
製備的聚合物p1在熱分析譜圖如圖1所示,從圖中可以看出,聚合物p1的分解溫度(失重5%時的溫度)為412℃。
實施例17
聚合物p2的製備
在氬氣氛圍下,將3,6-二(4,4,5,5-四甲基-1,3,2-二氧雜硼烷-二基)-n-辛基咔唑(145.01mg,272.92μmol)和菲並-s,s-二氧-而苯並噻吩(300mg,272.92μmol)加入100ml兩口瓶內,再加入8ml甲苯進行完全溶解,再加入醋酸鈀(2.45mg,10.92μmol)和三環己基膦(6.12mg,21.83μmol),然後加入2ml四乙基氫氧化銨,升溫至80℃,反應24小時;然後加入30mg苯硼酸進行封端,12小時後,再用0.1ml溴苯進行封端;繼續反應12小時之後,將產物滴加在甲醇中沉澱出來,攪拌,過濾,再將粗產物溶於20ml的甲苯中,以200~300目矽膠為固定相,用甲苯為洗脫劑進行柱層析,再將溶劑減壓濃縮後,再一次在甲醇中沉析出來,攪拌,過濾,真空乾燥後得到聚合物固體;最後再依次用甲醇、丙酮、四氫呋喃各抽提24小時,除去小分子;將濃縮後的四氫呋喃溶液滴入甲醇中沉析,真空乾燥後得到的纖維狀固體共軛聚合物p2。
製備的聚合物p2在薄膜狀態下的光致發光譜圖如2所示,從圖中可以看出,聚合物p2的最大發射峰位於556nm。
實施例18
聚合物p3的製備
在氬氣氛圍下,將茚芴硼酸(260.66mg,272.92μmol)和菲並-s,s-二氧-而苯並噻吩(300mg,272.92μmol)加入100ml兩口瓶內,再加入8ml甲苯進行完全溶解,再加入醋酸鈀(2.45mg,10.92μmol)和三環己基膦(6.12mg,21.83μmol),然後加入2ml四乙基氫氧化銨,升溫至80℃,反應24小時;然後加入30mg苯硼酸進行封端,12小時後,再用0.1ml溴苯進行封端;繼續反應12小時之後,將產物滴加在甲醇中沉澱出來,攪拌,過濾,再將粗產物溶於20ml的甲苯中,以200~300目矽膠為固定相,用甲苯為洗脫劑進行柱層析,再將溶劑減壓濃縮後,再一次在甲醇中沉析出來,攪拌,過濾,真空乾燥後得到聚合物固體;最後再依次用甲醇、丙酮、四氫呋喃各抽提24小時,除去小分子;將濃縮後的四氫呋喃溶液滴入甲醇中沉析,真空乾燥後得到的纖維狀固體共軛聚合物p3。
實施例19
基於小分子的電致發光器件的製備
在預先做好的方塊電阻為20ω/□的氧化銦錫(ito)玻璃上,先依次用丙酮,洗滌劑,去離子水和異丙醇超聲清洗,等離子處理10分鐘;在ito上旋塗參雜有聚苯乙烯磺酸的聚乙氧基噻吩(pedot:pss=1:1,w/w)膜,厚度為150nm;pedot:pss膜在真空烘箱裡80℃下乾燥8小時;隨後將雙極性小分子發光材料p1、p2、p3的氯苯溶液(1wt%)分別旋塗在pedot:pss膜的表面,厚度為80nm,作為發光層;最後在發光層上依次蒸鍍一薄層csf(1.5nm)和120nm厚的金屬al層。
聚合物p3基於器件結構:ito/pedot/eml/csf/al的電流密度-流明效率譜圖如圖3所示,從圖中可以看出,基於聚合物p3的器件的最大流明效率為1.87cd/a。
對得到的電致發光器件分別進行光電性能測試,結果如表1所示。
表1基於聚合物p1~p3得到的電致發光器件的光電性能指標
由表可知,聚合物p1、p2和p3基於器件結構:ito/pedot/eml/csf/al的最大流明效率依次為1.12cd/a、1.47cd/a和1.87cd/a。
上述實施例為本發明較佳的實施方式,但本發明的實施方式並不受上述實施例的限制,其它任何未背離本發明的精神實質與原理下所作的改變、修飾、替代、組合、簡化均應為等效的置換方式,都包含在本發明的保護範圍之內。