十二烷氧基苯基卟啉苯甲醯胺辛烷亞胺‑苝‑癸烷亞胺己氧基苯並菲三元化合物的合成方法與流程
2023-12-10 05:58:57 4
本發明涉及一種十二烷氧基苯基卟啉苯甲醯胺辛烷亞胺-苝-癸烷亞胺己氧基苯並菲三元化合物的合成方法
背景技術:
卟啉類分子是一種具有大π共軛體系的化合物,有著類似盤狀液晶分子的盤狀中心。同時卟啉盤狀液晶由於其芳香核中π-π軌道間的相互作用,能夠在一維方向上自組裝堆積成柱狀相,從而使電荷載流子沿著主體方向的遷移率要遠遠高於沿柱間方向的遷移。此外,高度對稱的盤狀液晶還有著較大的折射指數,較寬的相變行為和較高的焓變等特徵使得卟啉液晶的研究有著重要的意義。
苝醯亞胺類盤狀液晶材料是一類具有特殊稠環結構的化合物,具有良好的光、熱穩定性和抗氧化性,同時擁有較寬的光譜吸收範圍,以及高螢光量子產率等優良的光學性質。由於苝核呈缺電子體系性質,具有較高的電子親和能,這使得苝醯亞胺類衍生物成為一種典型的n型有機半導體材料,因此在有機光伏領域得到了廣泛研究。
本發明設計併合成了以烷氧基卟啉為電子給體(donor),柔性烷氧基鏈為橋體(bridge),苝單醯亞胺二酯單元為電子受體(acceptor),烷氧基苯並菲作為電子載體的有d-b-a型結構的三元化合物,這類化合物作為材料可以應用在有機光伏材料、液晶材料、有機太陽能電池、有機發光二極體等領域。
技術實現要素:
本發明的目的是提供一種十二烷氧基苯基卟啉苯甲醯胺辛烷亞胺-苝-癸烷亞胺己氧基苯並菲三元化合物的合成方法。
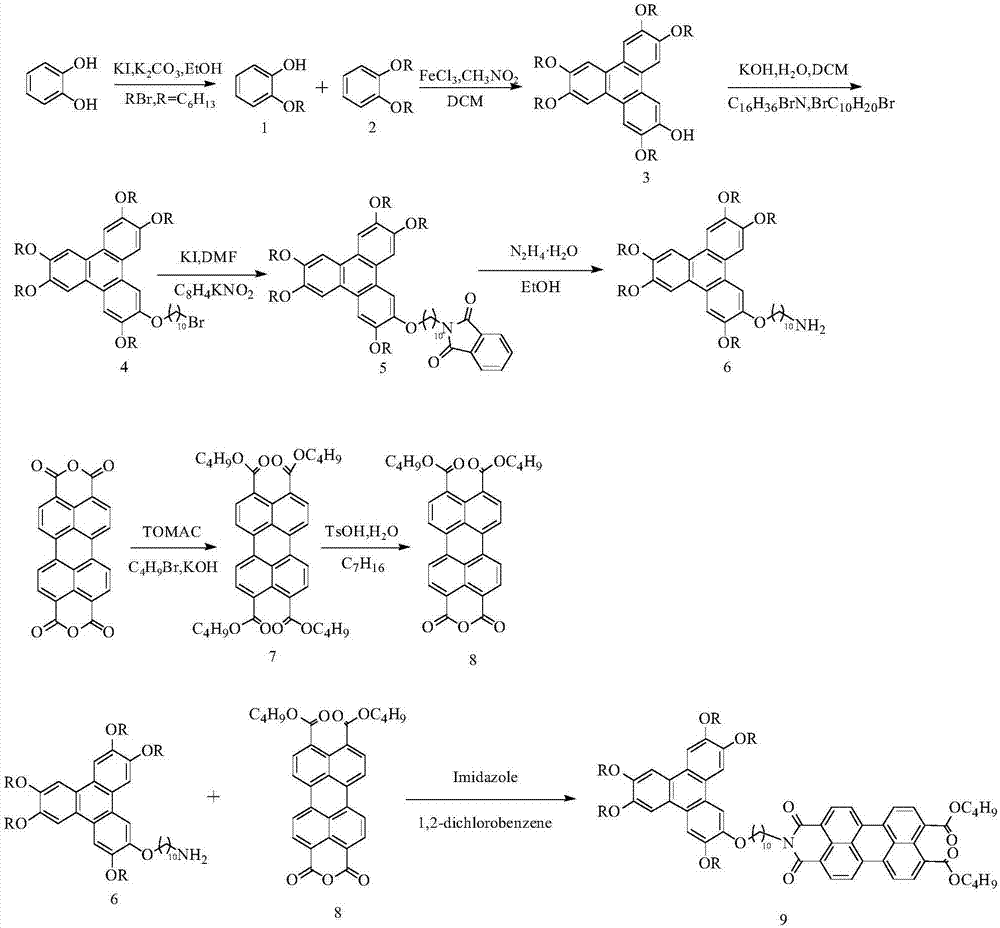
本發明合成路線分為以下五個部分:第一部分是以鄰苯二酚為原料得到3,6,7,10,11-五己氧基-2-癸伯胺苯並菲;第二部分將苝四甲酸二酐先水解後單邊縮合得到苝單酐二丁酯;第三部分是將上述得到的兩種化合物發生反應得到中間二聚體己氧基苯並菲癸烷亞胺橋連苝二丁酯;第四部分是通過一鍋法合成卟啉酯然後與1,8-辛二胺為主要原料合成一個帶氨基支鏈的卟啉化合物;第五部分是將上述第三、四部分得到的中間體在咪唑中發生醯胺反應合成得到目標化合物。
本發明設計併合成了以烷氧基卟啉為電子給體(donor),柔性烷氧基鏈為橋體(bridge),苝單醯亞胺二酯單元為電子受體(acceptor),烷氧基苯並菲作為電子載體的有d-b-a型結構的三元化合物,這類化合物作為材料可以應用在有機光伏材料、液晶材料、有機太陽能電池、有機發光二極體等領域。
附圖說明
圖1為十二烷氧基苯基卟啉苯甲醯胺辛烷亞胺-苝-癸烷亞胺己氧基苯並菲三元化合物的結構式。
圖2為合成十二烷氧基苯基卟啉苯甲醯胺辛烷亞胺-苝-癸烷亞胺己氧基苯並菲三元化合物的路線圖。
圖中標記為:1-化合物1,2-化合物2,3-化合物3,4-化合物4,5-化合物5,6-化合物6,7-化合物7,8-化合物8,9-化合物9,10-化合物10,11-化合物11,12-化合物12,13-化合物13。
具體實施方式
實施例:
實施例中所使用的化學試劑和溶劑均為分析純。
(1)鄰己氧基苯酚(化合物1)的合成:
將鄰苯2.2g二酚與溴3.96g代正己烷加到100ml的單口燒瓶中,再向燒瓶中加入少量的碘化鉀做催化劑,同時加入6.9g無水碳酸鉀和30ml的無水乙醇,室溫下攪拌1小時,然後加熱至80℃回流,恆溫反應24小時後結束反應。反應結束後對反應液進行萃取處理,先加入100ml的水,然後加入二氯甲烷30ml,收集有機層,在分次加入適量的二氯甲烷,直至新加入的二氯甲烷無色;重複該步驟三次,其中前兩次加入的為水,最後一次為飽和氯化鈉溶液。把收集的有機層用無水硫酸鈉乾燥,減壓旋幹得到的粗產物為液體。對粗產物進行蒸餾處理,收集溫度在140℃左右的餾分,得到純的化合物鄰己氧基苯酚1.67g,產率35%。1hnmr(300mhz,cdcl3)δ:6.95-6.82(m,4h),5.68(s,1h),4.02(m,2h),1.83-1.76(m,2h,j=6.6hz),1.48-1.31(m,6h),0.91(m,3h)。
(2)鄰二己氧基苯(化合物2)的合成
將2.2g鄰苯二酚與8.25g溴代正己烷加到100ml的單口燒瓶中,再向燒瓶中加入少量的碘化鉀做催化劑,同時加入6.9g無水碳酸鉀和30ml的無水乙醇,室溫下攪拌1小時,然後加熱至80℃回流,恆溫反應24小時後結束反應。反應結束後對反應液進行萃取處理,先加入100ml的水,然後加入二氯甲烷30ml,收集有機層,在分次加入適量的二氯甲烷,直至新加入的二氯甲烷無色;重複該步驟三次,其中前兩次加入的為水,最後一次為飽和氯化鈉溶液。把收集的有機層用無水硫酸鈉乾燥,減壓旋幹得到的粗產物為液體。對粗產物進行蒸餾處理,收集溫度在150℃左右的餾分,得到純的化合物鄰二己氧基苯5.34g,產率96%。1hnmr(300mhz,cdcl3)δ6.89(s,4h),3.99(m,4h),1.83-1.76(m,4h,j=7.0hz),1.49-1.31(m,12h),0.9(m,6hz)。
(3)單羥基五己氧基苯並菲單體3的合成
將1.16g化合物1和3.32g化合物2加入到滴液漏鬥中,取60ml二氯甲烷加入滴液漏鬥中,使化合物1和化合物2混合均勻;另取80ml二氯甲烷加入250ml的單口燒瓶中,並向其中加入三氯化鐵12.96g,打開攪拌,加入硝基甲烷8ml;冰水浴,保持溫度在0-5℃,滴加混合液,控制流速40分鐘滴完,反應2.5小時,反應過程中保持溫度不變。反應結束後加入30ml甲醇停止反應,再加入適量的水進行萃取,收集有機層,減壓旋幹,得到黑褐色固體粗產物。柱層層析法分離物質,淋洗液為石油醚:乙酸乙酯=40:1,純的單羥基五己氧基苯並菲為棕色液體,凝固後為紫色固體。1hnmr(400mhz,cdcl3)δ:7.96(s,1h),7.83(s,3h),7.82(s,1h),7.77(s,1h),5.91(s,1h),4.31-4.19(m,10h),1.98-1.90(m,10h),1.59-1.40(m,30h),0.96-0.92(m,15h);13cnmr(100mhz,cdcl3)149.12,148.99,148.79,148.72,145.82,145.23,123.92,123.61,123.54,123.19,122.95,107.58,107.39,107.29,107.22,106.42,104.31,69.91,69.86,69.59,69.09,31.68,31.64,31.62,29.46,29.40,29.27,25.85,25.82,22.65,22.61,14.05。
(4)溴癸烷氧基五己氧基苯並菲(化合物4)的合成
將2g化合物3和4.84g的1,10-二溴癸烷加入到100ml的單口燒瓶中,再加入0.26g
四丁基溴化銨;有機相的溶劑為二氯甲烷,無機相為水,且二氯甲烷和水的體積比為2:1。加入二氯甲烷30ml,水15ml,然後稱取氫氧化鉀0.6g加入體系,打開攪拌,室溫下反應24小時,觀察實驗進程,直至實驗結束。反應結束後進行萃取處理,先加入100ml的水,然後加入二氯甲烷30ml,收集有機層,在分次加入適量的二氯甲烷,直至新加入的二氯甲烷無色;重複該步驟三次,其中前兩次加入的為水,最後一次為飽和氯化鈉溶液。把收集的有機層用無水硫酸鈉乾燥,減壓旋幹得到的粗產物,柱層層析法分離提純,淋洗液為石油醚:二氯甲烷=5:1,最後得到2.52g化合物4。
(5)癸烷橋連五己氧基苯並菲、鄰苯二甲醯亞胺(化合物5)的合成
將2.52g化合物4和0.62g鄰苯二甲醯亞胺鉀加入到乾燥處理後的單口燒瓶中,然後再加入0.086g碘化鉀和dmf30ml,連接好冷凝裝置,打開攪拌,升溫至100℃反應8h,觀察實驗進程,直至反應結束。反應結束後對反應液進行萃取處理,先加入100ml的水,然後加入二氯甲烷30ml,收集有機層,在分次加入適量的二氯甲烷,直至新加入的二氯甲烷無色;重複該步驟三次,其中前兩次加入的為水,最後一次為飽和氯化鈉溶液。把收集的有機層用無水硫酸鈉乾燥,減壓旋幹得到的粗產物,柱層層析法分離提純,最後得到2.16g化合物5,產率為80.6%。
(6)3,6,7,10,11-五己氧基-2-癸伯胺苯並菲(化合物6)的合成
將2.16g化合物5加入到100ml的單口燒瓶中,然後加入85%的水合肼10ml,另外加入50ml的乙醇,打開攪拌,升溫至90℃中水解,反應2.5小時,反應結束後進行萃取,先加入100ml的水,然後加入二氯甲烷30ml,收集有機層,在分次加入適量的二氯甲烷,直至新加入的二氯甲烷無色;重複該步驟三次,其中前兩次加入的為水,最後一次為飽和氯化鈉溶液。把收集的有機層用無水硫酸鈉乾燥,減壓旋幹得到的粗產物,柱層層析法分離提純,淋洗液為二氯甲烷:甲醇=10:1,得到純的1.7g化合物6。
(7)苝四甲酸四丁酯(化合物7)的合成
配製質量分數為2.5%的氫氧化鉀水溶液,然後稱取3.8g的3,4,9,10-苝四羧酸酐加入氫氧化鉀水溶液中,在75℃下反應1.5小時,反應結束後過濾,收集濾液,向濾液中加入13.9g的1-溴代正丁烷和四丁基溴化銨,在100℃下反應2小時。反應結束後萃取,先加入100ml的水,然後加入二氯甲烷30ml,收集有機層,在分次加入適量的二氯甲烷,直至新加入的二氯甲烷無色;重複該步驟三次,其中前兩次加入的為水,最後一次為飽和氯化鈉溶液。把收集的有機層用無水硫酸鈉乾燥,減壓旋幹得到粗產物,用二氯甲烷/乙醇重結晶,過濾,得濾餅6.0g,產率94.8%。mp:162.6-163.8℃。
(8)苝單酐二甲酸二丁酯(化合物8)的合成
將6.0g化合物7加入到250ml的單口燒瓶中,然後向其中分別加入甲苯8ml和正庚烷40ml(甲苯和正庚烷的體積比為1:5),升溫至86℃攪拌1小時,然後再加入1.75g對甲苯磺酸,升溫至95℃,恆溫反應3小時;期間對實驗進程進行觀測,直至反應結束。反應結束後自然冷卻,減壓抽濾,得濾餅。然後用二氯甲烷/甲醇重結晶,重複三次,得到紅色固體4.36g,產率91%。
(9)癸烷亞胺五己氧基苯並菲-苝二甲酸二丁酯(化合物9)的合成
化合物6和化合物8按摩爾比1:1投入到單口燒瓶中,加入適量的咪唑做溶劑,升溫至130℃反應。反應持續5小時左右。反應結束後進行萃取,先加入100ml的水,然後加入二氯甲烷30ml,收集有機層,在分次加入適量的二氯甲烷,直至新加入的二氯甲烷無色;重複該步驟三次,其中前兩次加入的為水,最後一次為飽和氯化鈉溶液。把收集的有機層用無水硫酸鈉乾燥,減壓旋蒸得到紅色粗產物,柱層層析提純,得到目標產物中間體。
(10)對十二烷氧基苯甲醛(化合物10)的合成:
將2.44g對羥基苯甲醛,6.73g溴代十二烷和5.52g無水碳酸鉀,溶於20ml經cah2乾燥的n,n-二甲基甲醯胺溶劑。氮氣保護下,加熱至80℃攪拌12小時。冷卻至室溫,將反應液倒入100ml水中,用20ml二氯甲烷萃取,用無水硫酸鈉進行乾燥有機層,經旋轉蒸發儀除去溶劑後,用200-300目矽膠柱層析提純(淋洗液是二氯甲烷:石油醚體積比為1:5-1:2),得到淡黃色液體即化合物1(5.75g,產率98%)。bp>300℃,ir(kbr)νmax(cm-1):1610,1380,1260,841;1hnmr(300mhz,cdcl3)δ/ppm:9.87(s,h),7.68(t,j=6.9hz,2h),6.89(d,j=8.4hz,2h),3.99(t,j=6.9hz,2h),1.83-1.76(m,j=6.9hz,4h),0.96-1.31(m,j=5.4hz,10h).
(11)5-對苯甲酸甲酯基-10,15,20-三對十二烷氧基苯基卟啉(化合物11)的合成:
將5.88g化合物10,1.20g對醯基苯甲酸甲酯和1.85g間硝基苯甲酸,溶於32ml二甲苯溶液,通過恆壓滴液漏鬥滴加32ml含有1.91g吡咯的二甲苯溶液10分鐘,加熱140℃攪拌3.5小時。反應結束,冷卻至室溫,將180ml的無水甲醇倒入反應液中,靜置後抽濾,得到紫色物質,用200-300目矽膠柱層析提純(淋洗液是二氯甲烷/石油醚體積比為1:2-1:1),用二氯甲烷/無水甲醇=1:1重結晶,得到紫色固體即化合物2(2.3g,產率32.7%)。mp125.1-126.2℃,ir(kbr)νmax(cm-1):1720,1350,1240,803;1hnmr(400mhz,cdcl3)δ/ppm8.90(d,j=6.0hz,6h),8.76(d,j=4.7hz,2h),8.44(d,j=8.0hz,2h),8.30(d,j=8.0hz,2h),8.16-8.05(m,6h),7.29(d,j=8.4hz,6h),4.15(d,j=5.6hz,6h),4.11(s,3h),1.93(m,3h),1.77-1.54(m,12h),1.51-1.35(m,12h),1.06(m9h),1.00(t,j=6.8hz,9h),-2.74(s,2h).
(12)5-對苯甲醯胺辛胺基-10,15,20-三對十二烷氧基苯基卟啉(化合物12)合成
將1.3g化合物11加入到帶磁子攪拌的100ml的單口燒瓶中,氮氣保護,升溫至85℃,分別和至少3倍摩爾量的1,8-辛二胺反應,反應結束後對反應液進行萃取處理,先加入100ml的水,然後加入二氯甲烷30ml,收集有機層,在分次加入適量的二氯甲烷,直至新加入的二氯甲烷無色;重複該步驟三次,其中前兩次加入的為水,最後一次為飽和氯化鈉溶液。把收集的有機層用無水硫酸鈉乾燥,減壓旋蒸得到紅褐色粗產物。然後用柱層層析提純,分別得到目標產物12。
(13)十二烷氧基苯基卟啉苯甲醯胺辛烷亞胺-苝-癸烷亞胺己氧基苯並菲(化合物13)的合成
化合物9和化合物12按摩爾量1:1投入到100ml的單口燒瓶中,同時接入適量的鄰二氯苯和咪唑作為溶劑,氮氣保護;打開攪拌,升溫至140℃反應24小時,反應結束後經萃取、柱層層析分離提純得到純的化合物13。mp85.4-86.8℃,1hnmr(500mhz,cdcl3)δ8.89(m,2h),8.86–8.81(m,1h),8.71(d,j=4.5hz,1h),8.30(m,1h),8.19(d,j=8.0hz,1h),8.17–8.08(m,4h),8.06(d,j=8.5hz,2h),7.88(m,1h),7.79(m,1h),7.45(m,3h),7.24(m,2h),6.63(s,1h),-2.90(s,1h).
ir(kbr)νmax(cm-1):3420,2912,2852,1692,1658,1590,1506,1468,1434,1382,1347,1246,1177,1035,967,845,798,737,629.