一種高比電容的碳組合材料及其製備方法和應用與流程
2023-05-20 09:55:06 1
本申請涉及碳電極材料領域,特別是涉及一種高比電容的碳組合材料及其製備方法和應用。
背景技術:
超級電容器憑藉其快速的鋰離子傳輸性能得到了廣泛的研究。和鋰離子二次電池相比,超級電容具有超高的功率密度和循環穩定性,更是受到學術界和工業界的青睞。根據儲鋰機制超級電容器可以分為兩類,雙電層電容和贗電容。
碳材料因導電性高、比表面積大,而且碳的前驅體原料豐富等優勢,被廣泛應用於超級電容器。為了獲得高的電容值,碳電極材料必須具備三個特點,第一高比表面積,第二合適的孔徑分布,第三雜原子摻雜。其中,高比表面積和孔徑分布,已經有相當多的研究,例如通過微波法、煅燒法、酸鹼腐蝕法等製備高比表面積、孔徑分布均勻的碳材料。對於原子摻雜方面,現有的研究顯示,原子摻雜可以增加碳材料的贗電容,但是會降低其導電性,因此,必須要在導電性和贗電容之間尋求平衡,以獲得綜合性能良好的碳電極材料。
技術實現要素:
本申請的目的是提供一種新的高比電容的碳組合材料及其製備方法和應用。
本申請採用了以下技術方案:
本申請的一方面公開了一種高比電容的碳組合材料,該碳組合材料由原子摻雜量為10%-25%的第一組分碳材料,與原子摻雜量為0%-10%的第二組分碳材料混合而成。
需要說明的是,本申請中,第一組分碳材料由於具有比較高含量的原子摻雜,其贗電容相對較高,但其導電性差;第二組分碳材料的原子摻雜量低,導電性較好。本申請的碳組合材料,其關鍵就在於將高贗電容的第一組分碳材料,與高導電性的第二組分碳材料混合,將兩種具有不同優勢的碳材料混合,使其優勢互補,進而提高了碳組合材料整體的比電容值。其中,第二組分碳材料中原子摻雜的量為0%-10%,當原子摻雜的量為0時,表示採用的是沒有進行原子摻雜的碳材料。還需要說明的是,第二組分碳材料的原子摻雜的量為10%時,第一組分碳材料的原子摻雜的量是大於10%的,也就是說,第一組分碳材料和第二組分碳材料兩者的原子摻雜的量不同。
優選的,原子摻雜可重複的選自氮摻雜、氧摻雜、硫摻雜和磷摻雜的至少一種。
優選的,第一組分碳材料與第二組分碳材料的重量比為2:8-8:2。
需要說明的是,根據本申請的發明思路,只要將高原子摻雜的第一組分碳材料和低原子摻雜的第二組分碳材料混合,即可獲得比電容改善的碳組合材料;但是,考慮到比電容改善效果,本申請優選的方案中特別限定第一組分碳材料與第二組分碳材料的重量比為2:8-8:2。
本申請的另一面公開了本申請的碳組合材料在超級電容器中的應用。
本申請的另一面公開了一種由本申請的碳組合材料製備的極片。
本申請的另一面公開了一種採用本申請的極片的超級電容器。
可以理解,本申請提供的高比電容的碳組合材料,本身就是針對超級電容器而研究的,因此,將其製成極片用於超級電容器,能夠有效的提高超級電容器的電容性能。
本申請的再一面公開了一種本申請的碳組合材料的製備方法,包括以下步驟,
(a)將碳源與普魯士藍作為原材料,或者含摻雜原子的碳源與醋酸鐵作為原材料,分散到水和乙醇的混合溶液中,研磨混勻,將研磨產物烘乾,作為混合原料;將混合原料在氮氣氛下煅燒,煅燒條件為,2-5℃/min的速度升溫至450℃-650℃,保溫4-10h;煅燒完成後自然冷卻,然後,使煅燒產物暴露於空氣中,使其自燃燒;採用濃酸溶液對自燃燒的產物進行洗滌,洗滌過程中邊攪拌邊加熱,加熱溫度為60℃-90℃;洗滌完後離心乾燥,獲得原子摻雜量為10%-25%的所述第一組分碳材料;
(b)將步驟(a)製備的第一組分碳材料,在氬氣中,且大於650℃的溫度下煅燒6h,獲得原子摻雜量為0%-10%的所述第二組分碳材料;
(c)將步驟(a)製備的第一組分碳材料,與步驟(b)製備的第二組分碳材料混合,即獲得所述碳組合材料。
需要說明的是,步驟(a)中,乙醇和水的混合溶液中,乙醇主要起到分散作用,通常按照水:乙醇體積比1:5左右即可。
還需要說明的是,步驟(a)中,煅燒條件中煅燒溫度450℃-650℃,不同的煅燒溫度,獲得的碳材料具有不同的導電性和原子摻雜含量,總的來說溫度越高,摻雜原子含量相對較低,導電性較強;至於煅燒的時間,即保溫4-10h,主要是為了保障煅燒充分,保障材料的一致性。
優選的,碳源為葡萄糖,含摻雜原子的碳源為聚苯胺、三聚氰胺、海藻酸鈉、噻吩和肌醇六磷酸中的至少一種。
需要說明的是,本申請採用的鐵基化合物中,普魯士藍還有一個特殊的作用,即提供氮源,因此,在本申請的一種實現方式中,直接採用葡萄糖作為碳源,而不需要其它的含摻雜原子的碳源,與普魯士藍混合、研磨、煅燒即可獲得氮摻雜的碳材料。而採用含摻雜原子的碳源時,為了不引入其它雜原子,鐵基化合物採用醋酸鐵,這樣可以獲得含摻雜原子的碳材料;其中,聚苯胺和三聚氰胺為含摻雜原子氮的碳材料,海藻酸鈉為含摻雜原子氧的碳材料,噻吩為含摻雜原子硫的碳材料,肌醇六磷酸為含摻雜原子磷的碳材料。
優選的,步驟(a)中,濃酸溶液為濃硫酸。其中濃酸溶液洗滌是為了去除碳材料中的鐵原子,從而獲得比表面積大的碳殼材料。
優選的,步驟(b)中,在氬氣中煅燒的溫度為850℃-1050℃。
需要說明的是,步驟(b)中,煅燒溫度越高,摻雜原子的含量越低,本申請的實現方式中採用調控煅燒溫度控制摻雜原子含量,這只是摻雜原子含量的一種調控途徑,不排除可以採用實驗室使用的其它調控摻雜原子含量的方法。
本申請的有益效果在於:
本申請的碳組合材料中,第一組分碳材料的原子摻雜量相對較高,具有高贗電容的特點,而第二組分碳材料的原子摻雜量相對較低,具有較好的導電性,兩者優化互補;因此,將本申請的碳組合材料製成極片,在脫嵌鋰的過程中,第二組分碳材料不僅提供了導電網絡而且可以提供一部分贗電容,第一組分碳材料在導電網絡裡面更有助於摻雜原子贗電容的發揮,兩種碳材料相互作用,從而提高了電極材料的整體性能。
附圖說明
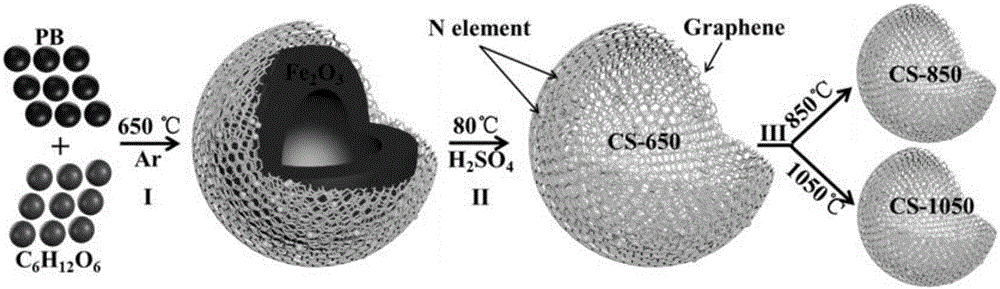
圖1是本申請實施例中製備碳材料的工藝流程示意圖;
圖2是本申請實施例中第一組分碳材料和第二組分碳材料相互作用的結構示意圖;
圖3是本申請實施例中製備的碳材料CS-650的透射電鏡圖;
圖4是本申請實施例中製備的碳材料CS-850的透射電鏡圖;
圖5是本申請實施例中三種碳材料製備成極片的倍率性能對比圖,其中「■」曲線為碳材料CS-650的倍率性能曲線,「●」曲線為碳材料CS-850的倍率性能曲線,「▲」曲線為碳材料CS-650和CS-850組成的碳組合材料的倍率性能曲線;
圖6是本申請實施例中製備的碳材料CS-1050的透射電鏡圖;
圖7是本申請另外一個實施例中三種碳材料製備成極片的倍率性能對比圖,其中「■」曲線為碳材料CS-650的倍率性能曲線,「●」曲線為碳材料CS-1050的倍率性能曲線,「▲」曲線為碳材料CS-650和CS-1050組成的碳組合材料的倍率性能曲線。
具體實施方式
本申請的發明人在大量的試驗和研究過程中發現,原子摻雜可以增加碳材料的贗電容,進而提高電極材料的整體性能;但是,原子摻雜量的增加,相應的會減小其導電性。因此,如何同時獲得高贗電容和高導電性的碳材料,這對本領域的研究人員來說是一個無法逾越的矛盾。本申請經過研究證實,將具有高贗電容的原子摻雜量高的碳材料,與具有高導電性的原子摻雜量低的碳材料混合,作為碳組合材料,可以產生1+1大於2的比電容和倍率性能。
本申請的碳組合材料的製備方法,如圖1所示,先將原材料混合,煅燒形成複合材料,然後經過酸洗,去除鐵的氧化物,即獲得原子摻雜量高的第一組分碳材料,再進一步的,對第一組分碳材料進行不同溫度的高溫煅燒,即獲得原子摻雜量低的第二組分碳材料,圖1所示的是本申請的兩種實現方式中,分別採用850℃和1050℃的煅燒溫度對原子摻雜量高的第一組分碳材料進行煅燒,獲得原子摻雜量低的第二組分碳材料。
本申請的碳組合材料中,第一組分碳材料的原子摻雜量相對較高,具有高贗電容的特點,而第二組分碳材料的原子摻雜量相對較低,具有較好的導電性,兩者優化互補,如圖2所示,圖2中A表示原子摻雜量低的第二組分碳材料,B表示原子摻雜量高的第一組分碳材料,電子主要通過第二組分碳材料進行傳遞,而第一組分碳材料主要發揮高贗電容作用。
下面通過具體實施例對本申請作進一步詳細說明。以下實施例僅對本申請進行進一步說明,不應理解為對本申請的限制。
實施例一
本例以葡萄糖為碳源製備碳組合材料,具體製備方法如下:
(a)稱取0.06g葡萄糖置於研缽中,滴加1mL水和5mL乙醇,製成葡萄糖漿料;稱取0.3g普魯士藍,即亞鐵氰化鐵,將其加入所製備的葡萄糖漿料中研磨,然後置於80℃鼓風乾燥箱中4小時;取出乾燥後的混合原料,在氮氣環境下煅燒,煅燒條件為:以2℃/min的速度升溫,在650℃下保溫6h;煅燒完成後,自然冷卻至室溫,然後從石英管中取出煅燒後的樣品,在空氣中震蕩,使其充分進行自燃燒;在空氣中震蕩約30min後,將得到的樣品倒入濃硫酸中,在攪拌的環境下加熱80℃,保持12小時,最後,將樣品進行離心洗滌乾燥,乾燥溫度為80℃,即獲得本例的氮摻雜量高的第一組分碳材料,命名為CS-650。
(b)將碳材料CS-650在850℃進行煅燒,煅燒整個過程在氬氣中進行,保溫6個小時,得到本例的氮摻雜量低的第二組分碳材料,命名為CS-850。
(c)本例將步驟(a)製備的第一組分碳材料,與步驟(b)製備的第二組分碳材料,按照5:5的重量比混合,即獲得本例的碳組合材料,命名為CS-650+850。
採用透射電鏡對本例製備的碳材料CS-650和CS-850進行觀察,結果分別如圖3和圖4所示,圖4中可見,提高煅燒溫度後獲得的碳材料CS-850有carbon layers出現,石墨化程度更高,導電性更好;而圖3的碳材料CS-650則沒有carbon layers出現。
同時,採用X射線光電子能譜分析測定本例製備的碳材料CS-650和CS-850的氮摻雜含量,結果顯示,CS-650的氮摻雜含量為12.8%,CS-850的氮摻雜含量為9.1%。
將本例製備的碳組合材料CS-650+850製備成極片,具體製備方法如下:
稱取0.04g的CS-650和0.04g的CS-850,進行研磨混合,然後加入0.01g炭黑再次進行研磨混合,之後加入0.025g濃度40%的PTFE和2mL異丙醇進行充分混合,製造成膜,然後通過滾壓裁片製成電極。
用電化學工作站測試CV,用Maccor測試櫃測試不同電流密度下的電容值。CV測試電壓區間為-1.0V-0V,掃描速率10-100mVs-1。充放電曲線的電壓區間為-1.0V-0V,電流密度為0.2-50A g-1。測試結果如圖5所示。與此同時,作為對比,本例分別採用碳材料CS-650和碳材料CS-850,替換碳組合材料CS-650+850製備極片,並對其進行倍率性能測試。測試結果如圖5所示。
如圖5的結果顯示,根據充放電曲線得到了三種材料的倍率性能,顯然CS-650+850的電容倍率特性最好,在50A g-1的條件下獲得的比電容為108F g-1。然而CS-650和CS-850的電容值為10F g-1和93F g-1。可見,本例將原子摻雜量高的第一組分碳材料和原子摻雜量低的第二組分碳材料混合製成的CS-650+850碳組合材料獲得了比兩者單獨使用更高的比電容。
實施例二
本例在步驟(b)中,將碳材料CS-650在1050℃進行煅燒,獲得的原子摻雜量低的第二組分碳材料命名為CS-1050。其餘與實施例一相同。
同樣的,採用透射電鏡對本例製備的碳材料CS-1050進行觀察,結果如圖6所示,可見,與碳材料CS-650相比,提高煅燒溫度後獲得的碳材料CS-1050有carbon layers出現,石墨化程度更高,導電性更好。
採用X射線光電子能譜分析測定本例製備的碳材料CS-1050的氮摻雜含量,結果顯示,CS-1050的氮摻雜含量為3.3%。
採用實施例一相同的方法將本例的碳組合材料CS-650+1050製備成極片,並以碳材料CS-1050為對比也製成極片,分別對本例的兩個極片以及實施例一的碳材料CS-850製成的極片進行倍率性能測試,測試結果如圖7所示,根據充放電曲線得到了三種材料的倍率性能,顯然CS-650+1050的電容倍率特性最好,在50A g-1的條件下獲得的比電容為100F g-1。然而CS-650和CS-850的電容值為10F g-1和46F g-1。
實施例三
本例採用含氧原子的碳源海藻酸鈉製備碳組合材料,具體方法如下:
(a)稱取0.06g海藻酸鈉置於研缽中,滴加1ml水和5ml乙醇,製成漿料;稱取0.3g醋酸鐵,將其加入所製備的漿料中研磨,然後置於80℃鼓風乾燥箱中4小時;取出乾燥後的混合原料,在氮氣環境下煅燒,煅燒條件為:以2℃/min的速度升溫,在550℃下保溫6h;煅燒完成後,自然冷卻至室溫,將得到的樣品倒入濃硫酸中,在攪拌的環境下加熱80℃,保持12小時,最後,將樣品進行離心洗滌乾燥,乾燥溫度為80℃,即獲得本例的氧摻雜量高的第一組分碳材料,命名為HZSN-550。
(b)將HZSN-550在850℃進行煅燒,煅燒整個過程在氬氣中進行,保溫6個小時,得到氧摻雜量低的第二組分碳材料,命名為HZSN-850。
(c)本例將步驟(a)製備的第一組分碳材料,與步驟(b)製備的第二組分碳材料,按照5:5的重量比混合,獲得本例的碳組合材料,命名為HZSN-550+850。
採用透射電鏡對本例製備的碳材料HZSN-550和HZSN-850進行觀察,結果顯示,提高煅燒溫度後獲得的碳材料HZSN-850有carbon layers出現,石墨化程度更高,導電性更好。採用X射線光電子能譜分析測定本例製備的碳材料HZSN-550和HZSN-850的氧摻雜含量,結果顯示,HZSN-550的氧摻雜含量為20%,HZSN-850的氧摻雜含量為5%。
採用實施例一相同的方法製備極片,並用電化學工作站測試CV,用Maccor測試櫃測試不同電流密度下的電容值。並且,作為對比,分別以碳材料HZSN-550和HZSN-850製備極片,測試其倍率性能。結果顯示,根據充放電曲線得到了三種材料的倍率性能,顯然HZSN-550+850的電容倍率特性最好,在50A g-1的條件下獲得的比電容為120F g-1。然而HZSN-550和HZSN-850的電容值分別為100F g-1和60F g-1。可見,本例將氧原子摻雜量高的第一組分碳材料和氧原子摻雜量低的第二組分碳材料混合製成的HZSN-550+850碳組合材料獲得了比兩者單獨使用更高的比電容。
實施例四
本例採用含硫原子的碳源噻吩製備碳組合材料,具體方法如下:
(a)稱取0.06g噻吩置於研缽中,滴加1ml水和5ml乙醇,製成漿料;稱取0.3g醋酸鐵,將其加入所製備的漿料中研磨,然後置於80℃鼓風乾燥箱中4小時;取出乾燥後的混合原料,在氮氣環境下煅燒,煅燒條件為:以2℃/min的速度升溫,在550℃下保溫6h;煅燒完成後,自然冷卻至室溫,將得到的樣品倒入濃硫酸中,在攪拌的環境下加熱80℃,保持12小時,最後,將樣品進行離心洗滌乾燥,乾燥溫度為80℃,即獲得本例的硫摻雜量高的第一組分碳材料,命名為HZSN-550。
(b)將SF-550在850℃進行煅燒,煅燒整個過程在氬氣中進行,保溫6個小時,得到硫摻雜量低的第二組分碳材料,命名為SF-850。
(c)本例將步驟(a)製備的第一組分碳材料,與步驟(b)製備的第二組分碳材料,按照5:5的重量比混合,獲得本例的碳組合材料,命名為SF-550+850。
採用透射電鏡對本例製備的碳材料SF-550和SF-850進行觀察,結果顯示,提高煅燒溫度後獲得的碳材料SF-850有carbon layers出現,石墨化程度更高,導電性更好。採用X射線光電子能譜分析測定本例製備的碳材料SF-550和SF-850的硫摻雜含量,結果顯示,SF-550的硫摻雜含量為17%,SF-850的硫摻雜含量為3%。
採用實施例一相同的方法製備極片,並用電化學工作站測試CV,用Maccor測試櫃測試不同電流密度下的電容值。並且,作為對比,分別以碳材料SF-550和SF-850製備極片,測試其倍率性能。結果顯示,根據充放電曲線得到了三種材料的倍率性能,顯然SF-550+850的電容倍率特性最好,在50A g-1的條件下獲得的比電容為140F g-1。然而SF-550和SF-850的電容值為120F g-1和80F g-1。可見,本例將硫原子摻雜量高的第一組分碳材料和硫原子摻雜量低的第二組分碳材料混合製成的SF-550+850碳組合材料獲得了比兩者單獨使用更高的比電容。
實施例五
本例採用含磷原子的碳源肌醇六磷酸製備碳組合材料,具體方法如下:
(a)稱取0.06g肌醇六磷酸置於研缽中,滴加1ml水和5ml乙醇,製成漿料;稱取0.3g醋酸鐵,將其加入所製備的漿料中研磨,然後置於80℃鼓風乾燥箱中4小時;取出乾燥後的混合原料,在氮氣環境下煅燒,煅燒條件為:以2℃/min的速度升溫,在550℃下保溫6h;煅燒完成後,自然冷卻至室溫,將得到的樣品倒入濃硫酸中,在攪拌的環境下加熱80℃,保持12小時,最後,將樣品進行離心洗滌乾燥,乾燥溫度為80℃,即獲得本例的磷摻雜量高的第一組分碳材料,命名為JC-550。
(b)將SF-550在850℃進行煅燒,煅燒整個過程在氬氣中進行,保溫6個小時,得到磷摻雜量低的第二組分碳材料,命名為JC-850。
(c)本例將步驟(a)製備的第一組分碳材料,與步驟(b)製備的第二組分碳材料,按照5:5的重量比混合,獲得本例的碳組合材料,命名為SF-550+850。
採用透射電鏡對本例製備的碳材料SF-550和SF-850進行觀察,結果顯示,提高煅燒溫度後獲得的碳材料JC-850有carbon layers出現,石墨化程度更高,導電性更好。採用X射線光電子能譜分析測定本例製備的碳材料JC-550和JC-850的磷摻雜含量,結果顯示,SF-550的磷摻雜含量為12%,SF-850的磷摻雜含量為3%。
採用實施例一相同的方法製備極片,並用電化學工作站測試CV,用Maccor測試櫃測試不同電流密度下的電容值。並且,作為對比,分別以碳材料JC-550和SF-850製備極片,測試其倍率性能。結果顯示,根據充放電曲線得到了三種材料的倍率性能,顯然JC-550+850的電容倍率特性最好,在50A g-1的條件下獲得的比電容為102F g-1。然而JC-550和JC-850的電容值為80F g-1和40F g-1。可見,本例將磷原子摻雜量高的第一組分碳材料和磷原子摻雜量低的第二組分碳材料混合製成的SF-550+850碳組合材料獲得了比兩者單獨使用更高的比電容。
以上內容是結合具體的實施方式對本申請所作的進一步詳細說明,不能認定本申請的具體實施只局限於這些說明。對於本申請所屬技術領域的普通技術人員來說,在不脫離本申請構思的前提下,還可以做出若干簡單推演或替換,都應當視為屬於本申請的保護範圍。