作為用於丙交酯聚合物的聚合催化劑的包含至少一個Cp環的第IVB族過渡金屬催化劑的製作方法
2023-04-30 15:55:51 3
聚(乳酸)(PLA)由於它們的生物可降解性和生物相容性已在過去幾十年期間被強烈地研究。PLA具有多樣性的物理性質並且已經在醫學應用和組織工程例如用於受控制的藥物釋放的介質中被使用。丙交酯(LA)的通過單位點引發劑的開環聚合(ROP)對於具有受控制的分子量和窄的分子量分布的PLA是最有效的路線。在一個丙交酯分子中的兩個立體中心產生三種有區別的構型異構體(S,S)-LA、(L-LA);(R,R)-LA、(D-LA)以及(R,S)-LA、(內消旋)-LA。(S,S)-LA和(R,R)-LA的1:1混合物被稱為外消旋-LA。
對於引發丙交酯的開環聚合有用的金屬絡合物是已知的。
Wenshan Ren等人,Inorganic Chemistry Communications,30,(2013),26-28報導了苄基釷金屬茂[η5-1,3-(Me3C)2C5H3]2Th(CH2Ph)2(1)和[η5-1,2,4-(Me3C)3C5H2]2Th(CH2Ph)2(2)在溫和的條件下可以引發外消旋-丙交酯(外消旋-LA)的開環聚合(ROP)。500當量的丙交酯的完全轉化在5小時內在40℃下在二氯甲烷中以[外消旋-LA]=1.0mol L-1發生,並且分子量分布在全部的單體比引發劑的範圍內是非常窄的(約1.15),這指示單位點的催化劑體系。
Yalan Ning等人,Organometallics 2008,27,5632-5640報導了用於L-丙交酯(L-LA)和ε-己內酯(ε-CL)的開環聚合和鏈轉移聚合的四種中性二茂鋯雙(酯烯醇化物)和非二茂鋯雙(烷氧基)絡合物。
AJ Chmura等人,Chem.Commun.2008,1293發現,鋯胺三(酚鹽)醇鹽和鉿胺三(酚鹽)醇鹽是極其活性的,產生高度雜同立構的聚丙交酯(heterotactic polylactide)。
本發明基於不同類別的化合物的發現,所述化合物具有作為用於丙交酯單體的聚合的引發劑的用途。
本發明提供具有下式的化合物,
La M(OR1)b R2c Xd
其中
M是選自Ti、Zr以及Hf的金屬;
L是選自以下的配體:全甲基並環戊二烯(Pn*=C8Me6)、(氫)全甲基並環戊二烯(Pn*(H)=C8Me6H)、(氫)並環戊二烯(Pn(H)=C8H8)、環戊二烯(Cp=C5H5)、茚(C7H7)以及亞乙基橋接的茚或矽烷橋接的茚;
R1是1-6C烷基、被取代的或未被取代的苯基、或被取代的或未被取代的苯基亞烷基;
R2是Me或Et
X是滷素
a=1至3,b=1至3,c=0或1並且d=0、1、2或3;
及其二聚體。
上文提到的並環戊二烯具有以下示出的結構:
矽烷橋可以任選地被一系列烷基取代。SBI配體指的是二甲基矽烷橋接的茚基配體。
本發明還提供了本發明的化合物作為在丙交酯單體的聚合中的引發劑的用途。
本發明還另外提供了用於產生聚丙交酯的工藝,所述工藝包括使丙交酯單體與本發明的化合物接觸。
如上文所陳述的,本發明的化合物具有以下的通式,
La M(OR1)b R2c Xd
其中L、M、R1、R2、X、a、b、c以及d為如上文所定義的,及其二聚體。
M是選自鈦、鋯以及鉿的第IV族過渡金屬。根據一個優選的實施方案,M是鈦。根據另一個優選的實施方案,M是鋯。根據不同的實施方案,M是鉿。
L是選自以下的配體:全甲基並環戊二烯(Pn*)、(氫)全甲基並環戊二烯(Pn*(H))、(氫)並環戊二烯(Pn(H))、環戊二烯(Cp)、茚以及亞乙基橋接的茚(EBI)和二甲基矽烷橋接的茚(SBI)。根據一個優選的實施方案,配體基團L是全甲基並環戊二烯。根據不同的實施方案,當M是Zr時,配體基團L是茚或EBI。
本發明的化合物是基於配體基團L的金屬醇鹽絡合物、金屬酚鹽絡合物或苯基亞烷基氧化金屬絡合物(phenylalkyleneoxide metal complex)。被附接至金屬M的OR1基團的R1選自1-6C烷基、被取代的或未被取代的苯基以及被取代的或未被取代的苯基亞烷基。優選地,對於R1,1-6C烷基是叔丁基。優選地,當R1是苯氧基(phenyloxide group)時,其是式–C6H3(R3)2的二烷基苯氧基,其中R3是1-4C烷基,尤其是Me、iPr或tBu。根據優選的實施方案,R1選自2,6-二甲基苯基、2,6-二異丙基苯基以及2,6-二叔丁基苯基。
根據不同的實施方案,R1可以是如上文提及的被取代的或未被取代的苯基亞烷基。實例包括–CH2C6H5和–CH(Me)C6H5。
本發明的金屬絡合物可以包含被附接至金屬M的一個R2基團。如果存在,那麼R2選自Me和Et。如果R2在絡合物中存在,那麼其優選地是甲基。
本發明的金屬絡合物可以包含被附接至M的滷素基團X。優選地,如果存在,那麼X是Cl。典型地,如果X在絡合物中存在,那麼d的值是1或2。
根據優選的實施方案,本發明的化合物是為以下的(半)金屬茂絡合物:
η5-Pn*(H)Ti(OtBu)3;或
η5-Pn*(H)Zr(OtBu)3;或
η5-Pn*(H)Zr(O-CH2C6H5)3;或
η5-Pn*(H)Zr(O-S-CH(CH3)C6H5)3;或
外消旋-η5-Pn*(H)Zr(O-外消旋-CH(CH3)C6H5)3;或
η5-Pn*(H)Zr(O-2,6-Me-C6H3)3;或
η5-Pn*(H)Zr(O-2,6-iPr-C6H3)3;或
η5-Pn*(H)ZrCl2(O-2,6-tBu-C6H3);或
η5-Pn*(H)Hf(O-2,6-Me-C6H3)3;或
η5-Pn*(H)HfCl(O-2,6-iPr-C6H3)2;或
η5-Pn*(H)HfCl2(O-2,6-tBu-C6H3)。
上文化合物可以例如通過使η5-Pn*(H)SnMe3與對應的金屬氯化物例如TiCl4、ZrCl4或HfCl4在苯中在80℃下反應持續2-72小時並且然後與合適的醇鉀鹽、酚鉀鹽或苯基亞烷基氧化鉀在室溫下在苯或甲苯中反應來製備。然而,用於製備本領域已知的化合物的任何合適的工藝都可以被用於其製備。
根據不同的優選的實施方案,本發明的(半)金屬茂絡合物是
[(Pn*)Ti(O-2,6-Me-C6H3)Cl];或
[η8-(Pn*)Ti(O-2,4-tBu-C6H3)Cl];或
[η8-(Pn*)Ti(O-2,6-Me-C6H3)2];或
[η8-(Pn*)Ti(OtBu)Cl];或
[η8-(Pn*)Ti(OtBu)2;或
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)Cl2];或
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)3]。
上文化合物可以例如通過使一當量的[η8-(Pn*)TiCl(μ-Cl)]2與二當量的KOR(其中R=2,6-MeC6H3或2,4-tBuC6H3)在室溫下在甲苯中反應持續24-48小時來產生。然而,用於製備本領域已知的化合物的任何合適的工藝都可以被用於其製備。
根據又另外的優選的實施方案,本發明的(半)金屬茂絡合物是
[(EBI)Zr(O-2,6-Me-C6H3)Cl];或
[Ind2Zr(O-2,6-Me-C6H3)Me];或
[Ind2Zr(O-2,6-Me-C6H3)Cl];或
[Ind2Zr(O-2,6-Me-C6H3)2];或
[Cp2Zr(O-2,6-Me-C6H3)2];或
[Cp2Zr(O-2,6-Me-C6H3)Cl];或
[Cp2Zr(O-2,6-Me-C6H3)Me]。
上文EBI Zr化合物可以通過使化學計量的量的[K(O-2,6-Me-C6H3)]和外消旋-[(EBI)ZrCl2]在室溫下在甲苯中在攪拌下反應持續18小時來產生。用於製備[(Ind)2Zr(OtBu)Me]的修改的程序包括使化學計量的量的[(Ind)2ZrMe2]和叔丁醇在室溫下在甲苯中在攪拌下反應持續18小時,隨後是在真空中濃縮。[(Ind)2Zr(O-2,6-Me-C6H3)Me]可以通過使化學計量的量的[(Ind)2ZrMe]和2,6-二甲基苯酚在室溫下在甲苯中在攪拌下反應持續18小時、隨後在真空中濃縮來製備。然而,用於製備本領域已知的化合物的任何合適的工藝都可以被用於其製備。
本發明的化合物作為在丙交酯單體的聚合中的引發劑是有用的。因此,本發明還涉及如前文描述的化合物作為在丙交酯單體的聚合中的引發劑的用途。
根據另外的方面,本發明提供了用於產生聚丙交酯的工藝,所述工藝包括使丙交酯單體與如前文描述的化合物接觸。
在該工藝的優選的實施方案中,丙交酯單體是L-丙交酯並且產生的聚丙交酯是等規立構聚丙交酯。在該工藝的另一個優選的實施方案中,丙交酯單體是外消旋-丙交酯並且產生的聚丙交酯是無規立構聚丙交酯。
實驗細節I-涉及全甲基並環戊二烯
一般程序
所有有機金屬合成都在氮氣的惰性氣氛下利用標準Schlenk技術在雙重真空-進口氣體歧管(dual vacuum-inlet gas manifold)或Braun手套箱上進行。在必要時,溶劑被SPS乾燥體系(己烷、戊烷、甲苯)乾燥。氘代的NMR溶劑在使用之前,經NaK(苯-d6、甲苯-d8)或CaH2(氯仿-d1)乾燥、真空轉移並且冷凍-抽吸-解凍-脫氣三次。元素分析由Stephen Boyer先生在倫敦都市大學(London Metropolitan University)的元素分析服務部進行。NMR光譜使用楊氏分接頭NMR管(Young’s tap NMR tubes)被記錄在Varian Mercury VX-Works 300MHz光譜儀上。1H光譜和13C{1H}NMR光譜通過殘餘的質子溶劑峰(protio-solvent peak)作參考。叔丁醇鉀購自Sigma-Aldrich並且按原樣使用。L-丙交酯和外消旋-丙交酯購自Alfa Aesar並且在使用之前被重結晶並且升華(10-2毫巴,50℃)。[K(O-2,6-Me-C6H3)]和[K(O-2,4-tBu-C6H3)]通過在室溫下攪拌在THF中的雙(三甲基甲矽烷基)醯胺鉀與合適的醇來製備。
X射線結晶學
使用全氟聚醚油將晶體固定在玻璃纖維上,轉移至在衍射計上的測角計頭並且在冷的氮氣的流中使用Oxford Cryosystems CRYOSTREAM單元迅速冷卻至150K。數據收集使用Enraf-Nonium FR590KappaCCD衍射計、利用石墨-單色化的Mo KαX射線輻射來進行。強度數據使用DENZO-SMN包來處理。結構使用直接法程序SIR92來求解,並且使用CRYSTALS程序組對所有F2數據應用全矩陣最小二乘法求精來求精。
聚合程序
所有聚合在包含0.4mL的具有[LA]0=0.104M的初始丙交酯濃度的丙交酯的苯-d6溶液和0.1mL的催化劑的苯-d6溶液(具有用於確保[LA]0/[初始]0=50的濃度)的楊氏分接頭NMR管中進行。丙交酯轉化率隨後通過比較在1H NMR光譜中的PLA和丙交酯單體的甲烷信號的積分值來計算。進行給定的聚合的溫度在80℃至100℃的溫度範圍內變化並且在合適的部分中被註明。
本發明的主題的另外的優點和特徵可以結合附圖取自以下詳述,在附圖中:
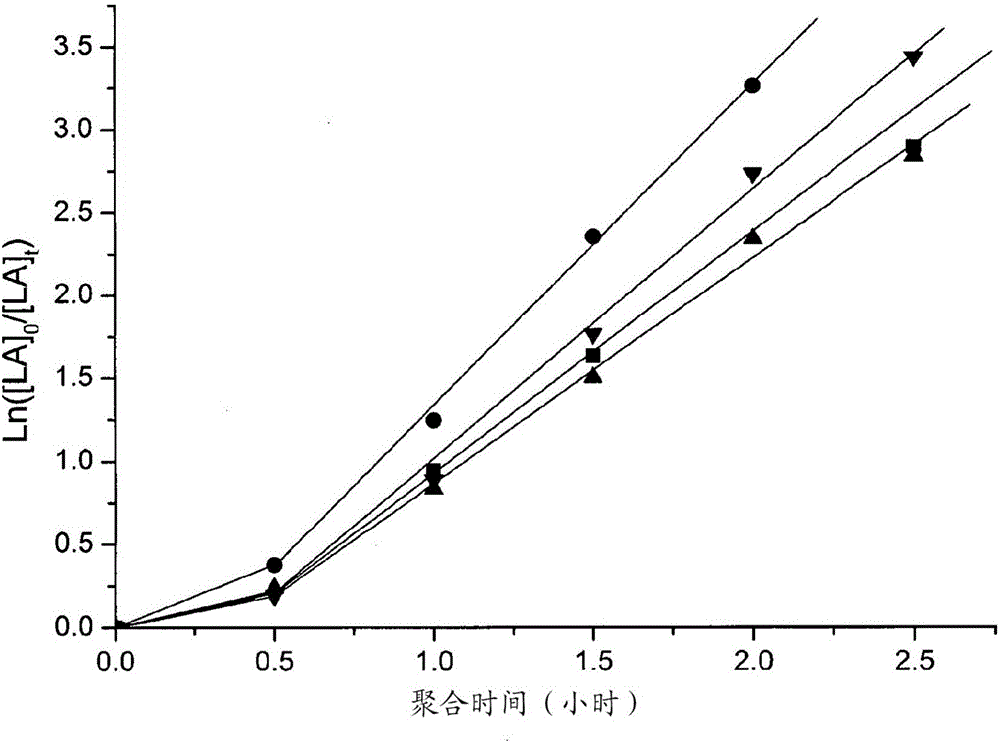
圖1:使用以下絡合物的L-丙交酯聚合:η5-Pn*(H)Ti(O-2,6-Me2C6H3)32(黑色正方形,k觀察=0.113±0.014h-1)、η5-Pn*(H)Zr(O-2,6-Me2C6H3)3 7(紅色圓形,k觀察=0.479±0.032h-1)、η5-Pn*(H)Zr(O-2,6-iPr2C6H3)3 8(粉色三角形,k觀察=0.391±0.022h-1)、η5-Pn*(H)ZrCl2(O-2,6-iBu2C6H3)9(深藍色三角形,k觀察=0.043±0.003h-1)、η5-Pn*(H)Hf(O-2,6-Me2C6H3)3 10(藍色三角形,k觀察=0.364±0.027h-1)、η5-Pn*(H)HfCl(O-2,6-iPr2C6H3)2 11(綠色菱形,k觀察=0.463±0.029h-1)以及η5-Pn*(H)HfCl2(O-2,6-iBuC6H3)12(紫色三角形,k觀察=0.086±0.020h-1)。聚合條件:氯仿-d1,在100℃下,其中[LA]0/[M]0=50,[LA]0=0.5M。
圖2:丙交酯聚合:L-丙交酯和S-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,5(黑色正方形,k觀察=1.166±0.068h-1);L-丙交酯和外消旋-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,6(紅色圓形,k觀察=1.954±0.063h-1);外消旋-丙交酯和外消旋-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,6(粉色三角形,k觀察=1.667±0.053h-1);外消旋-丙交酯和S-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,5(藍色三角形,k觀察=1.342±0.055h-1)。聚合條件:氯仿-d1,在80℃下,其中[LA]0/[M]0=50、[LA]0=0.5M。
圖3:丙交酯聚合:L-丙交酯和S-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,5(黑色正方形,k觀察=0.484±0.037h-1);L-丙交酯和外消旋-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,6(紅色圓形,k觀察=0.850±0.063h-1);外消旋-丙交酯和外消旋-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,6(粉色三角形,k觀察=0.767±0.037h-1);外消旋-丙交酯和S-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3,5(藍色三角形,k觀察=0.491±0.031h-1)。聚合條件:氯仿-d1,在60℃下,其中[LA]0/[M]0=50、[LA]0=0.5M。
圖4:使用S-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3 5的L-丙交酯聚合的艾林標繪圖(Eyring plot)。斜率=-5610±488,且R2=0.993。聚合條件:氯仿-d1,其中[LA]0/[Zr]0=50並且[LA]0=0.5M。
圖5:使用η5-Pn*(H)Zr(O-2,6-iPr2C6H3)3 8的L-丙交酯聚合:[LA]0/[Zr]0=25(黑色正方形,k觀察=0.521±0.021h-1);[LA]0/[Zr]0=50(紅色圓形,k觀察=0.391±0.022h-1);[LA]0/[Zr]0=100(藍色三角形,k觀察=0.377±0.015h-1);[LA]0/[Zr]0=200(粉色三角形,k觀察=0.235±0.004h-1)。聚合條件:氯仿-d1,在100℃下,其中[LA]0=0.5M。
圖6:使用η5-Pn*(H)Zr(O-2,6-iPr2C6H3)3 8的Ln(k觀察)相對於Ln([Zr]0)的標繪圖。斜率=0.350±82,R2=0.901。聚合條件:氯仿-d1,在100℃下,並且[LA]0=0.5M。
圖7:使用η5-Pn*(H)Zr(O-2,6-iPr2C6H3)3 8的L-丙交酯聚合:T=100℃(黑色正方形,k觀察=0.391±0.022h-1);T=90℃(紅色圓形,k觀察=0.151±0.008h-1);T=80℃(藍色三角形,k觀察=0.092±0.006h-1)。聚合條件:氯仿-d1,其中[LA]0/[M]0=50、[LA]0=0.5M。
圖8:使用η5-Pn*(H)Zr(O-2,6-iPr2C6H3)3 8的L-丙交酯聚合的艾林標繪圖。斜率=-9133±1881,且R2=0.959。聚合條件:氯仿-d1,其中[LA]0/[Zr]0=50並且[LA]0=0.5M。
圖9:L-丙交酯轉化率相對於時間的半對數標繪圖,[LA]0/[初始]0=50,[LA]0=0.104M,T=80℃,苯-d6(0.5mL),使用[η8-(Pn*)Ti(O-2,6-Me2C6H3)Cl](綠色點劃線)、[η8-(Pn*)Ti(O-2,4-tBu2C6H3)Cl](黑色短劃線)、[η8-(Pn*)Ti(O-2,6-Me2C6H3)2](紅色線)以及[η5-(Pn*H)Ti(O-2,6-Me2C6H3)Cl2](藍色點虛線)。省略誘導期。
圖10:丙交酯單體轉化率相對於時間的半對數標繪圖。省略誘導期。[LA]0=0.104M,[LA]0/[初始]0=50,T=90℃,苯-d6。使用[η8-(Pn*)Ti(O-2,6-Me2C6H3)2]、L-丙交酯(紅色線)和外消旋-丙交酯(黑色短劃線)的聚合。
圖11:L-丙交酯轉化率相對於時間的半對數標繪圖,[LA]0/[初始]0=50,[LA]0=0.50M,T=80℃,氯仿-d1(0.5mL),使用[(EBI)Zr(OC6H3Me2-2,6)Cl]1(點劃線)、[(Ind)2Zr(OtBu)Me]2(實線)、[(Ind)2Zr(OC6H3Me2-2,6)Me]3(短劃線)的聚合。
圖12:增長速率相對於溫度的倒數的半對數標繪圖,[LA]0/[2]0=50,[LA]0=0.50M,氯仿-d1,並且
圖13:丙交酯轉化率相對於時間的半對數標繪圖,[LA]0/[2]0=50,[LA]0=0.50M,T=80℃,氯仿-d1(0.5mL);L-丙交酯(實線),外消旋-丙交酯(短劃線)。
實施例1
[(Pn*)Ti(O-2,6-Me-C6Me2)Cl]的合成將[η8-(Pn*)TiCl(μ-Cl)]2(150mg,0.26mmol)和[K(O-2,6-MeC6H3)](78mg,0.50mmol)在甲苯(30mL)中合併並且留下在室溫下攪拌持續25小時。將產生的溶液過濾,然後在真空中濃縮。在-35℃下儲存甲苯濃縮的溶液持續24小時之後獲得了X射線質量單晶(X-ray quality single crystal)。收率=52%。1H NMR(苯-d6,25℃,300MHz):δ7.04(d,2H,3JHH=7.3Hz,Ar-H),6.82(t,1H,3JHH=7.4Hz,Ar-H),2.11,2.06,1.68(s,每6H,Pn-CH3),1.61(s,6H,Ar-CH3)。13C{1H}NMR(甲苯-d8,25℃,75.1MHz):δ130.69,126.16,14.36,123.41,120.41(季碳),18.09(Ar-CH3)13.20,12.47,11.11(Pn-CH3)。四級橋頭碳原子沒有觀察到並且某些季碳信號被溶劑共振遮蔽。對於C22H27ClTiO(%)的分析計算值:C,67.62,H,6.98;實測值:C,67.70;H,7.03。
實施例2
[η8-(Pn*)Ti(O-2,4-tBu-C6H3)Cl]的合成將[η8-(Pn*)TiCl(μ-Cl)]2(150mg,0.25mmol)和[K(O-2,4-tBu-C6H3)](120mg,0.50mmol)在甲苯(30mL)中在室溫下攪拌持續48小時。將產生的溶液過濾並且在真空中除去溶劑。隨後溶解在最少的熱的苯中並且在室溫下儲存24小時導致X射線質量單晶的形成。收率=55%。1H NMR(苯-d6,25℃,300MHz):δ7.54(d,1H,4JHH=2.4Hz,間位-Ar-H),7.18(dd,1H,3JHH=8.1Hz,4JHH=2.4Hz,間位-Ar-H),6.41(d,1H,3JHH=8.3Hz,鄰位-Ar-H),2.07,1.80,1.64(s,每6H,Pn-CH3),1.59(s,9H,C(CH3)),1.35(s,9H,C(CH3))。13C{1H}NMR(苯-d6,25℃,75.1MHz):δ161.21(本位-Ar),142.66,140.76,139.1,0 135.5,7 130.28,124.62(季碳),123.82,123.77(間位-Ar)122.82(季碳)121.22(鄰位-Ar),35.55,34.55(Ar-CM3),31.95,30.88(Ar-CH3),13.04,12.48,10.84(Pn-CH3)。對於C28H39TiClO(%)的分析計算值:C,70.8;H,8.29;實測值:C,70.65;H,8.23。
實施例3
[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]的合成將[η8-Pn*TiCl(μ-Cl)]2(150mg,0.25mmol)和[K(O-2,6-Me-C6H3)](160mg,0.50mmol)在甲苯(30mL)中在25℃下攪拌持續24小時。將產生的溶液過濾並且隨後在真空中濃縮。在-35℃下儲存甲苯濃縮的溶液持續24小時之後獲得了X射線質量單晶。收率=62%。1H NMR(苯-d6,25℃,300MHz):δ6.96(d,4H,3JHH=7.5Hz,間位-Ar-H),6.69(t,2H,3JHH=7.3Hz,對位-Ar-H),2.13(s,12H,Pn-CH3),1.81(s,12H,Ar-CH3),1.77(s,6H,Pn-CH3)。13C{1H}NMR(苯-d6,25℃,75.1MHz):δ162.52(本位-Ar),139.73 130.97 125.22 120.47 118.89(季碳),17.73(Ar-CH3),11.90 10.89(Pn-CH3)。四級橋頭碳原子沒有觀察到。對於C30H36TiO2(%)的分析計算值:C,75.62;H,7.63;實測值:C,75.48;H,7.77。
實施例4
[η8-(Pn*)Ti(OtBu)Cl]的合成將[η8-(Pn*)TiCl(μ-Cl)]2(150mg,0.25mmol)和[K(OtBu)](53mg,0.50mmol)在甲苯(30mL)中合併並且留下在室溫下攪拌持續2小時。將產生的溶液過濾並且在減壓下除去溶劑。添加最少的熱的己烷並且在冷卻至室溫並且在-35℃下儲存24小時之後獲得了X射線質量單晶。收率=54%。1H NMR(苯-d6,25℃,300MHz):δ2.09,1.90,1.62(s,每6H,Pn-CH3),1.31(s,9H,C(CH3))。13C{1H}NMR(苯-d6,25℃,75.1MHz):δ138.41,136.38(Pn-橋頭),128.86,122.16,121.91(Pn),81.16(C(CH3)),32.48(C(CH3)),13.09,12.85,10.86(Pn-CH3)。
實施例5
[η8-(Pn*)Ti(OtBu)2]的合成將[η8-(Pn*)TiCl(μ-Cl)]2(20mg,0.035mmol)和[K(OtBu)](16mg,0.14mmol)在苯-d6(1mL)中合併。將獲得的深紅色溶液過濾並且允許經歷緩慢蒸發,這導致X射線質量單晶的形成。1H NMR(苯-d6,25℃,300MHz):δ2.08(s,12H,Pn-CH3),1.76(s,6H,Pn-CH3),1.31(s,18H,C(CH3))。13C{1H}NMR(苯-d6,25℃,75.1MHz):134.38,128.71,116.58(Pn)76.44(C(CH3))33.60(C(CH3))13.07,10.81(Pn-CH3)。四級橋頭碳原子沒有觀察到。
實施例6
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)Cl)2]的合成將[η8-Pn*TiCl(μ-Cl)]2(350mg,0.57mmol)和[H(O-2,6-Me-C6H3)](139mg,1.14mmol)在甲苯(50mL)中在85℃下攪拌持續24小時,然後過濾並且在真空下濃縮。在-35℃下儲存濃縮的溶液持續24小時之後形成X射線質量單晶。收率=68%。1HNMR(苯-d6,25℃,300MHz):δ6.82-6.72(m,3H,Ar-H),3.19(q,1H,3JHH=7.3Hz,Pn-H),2.24(s,6H,Ar-CH3),2.23,2.12,2.08,1.79,1.44(s,每3H,Pn-CH3),0.84(d,3H,3JHH=7.5Hz,Pn-CH3)。13C{1H}NMR(苯-d6,25℃,75.1MHz)δ162.73(本位-Ar),150.62,147.79,141.18,136.86,129.80,129.07(季碳),128.13(間位-Ar)123.70(季碳)123.06(對位-Ar),44.19(sp3Pn),17.22(Ar-CH3),15.53,14.08,13.53,13.23,11.75,11.59(Pn-CH3)。四級橋頭碳原子沒有觀察到。對於C22H28Cl2TiO(%)的分析計算值:C,61.84;H,6.62;實測值:C,61.71;H,6.70。
實施例7
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)3]的合成將[η5-(Pn*H)Ti(OC6H3Me2-2,6)Cl2](50mg,0.12mmol)和[K(O-2,6-Me2C6H3)](37.5mg,0.24mmol)在甲苯(10mL)中的溶液在100℃下攪拌持續30分鐘。將產生的亮橙色溶液過濾並且在減壓下除去溶劑。隨後溶解在最少的熱的己烷中、隨後在-35℃下儲存持續24小時導致X射線質量單晶的形成。收率=65%。1H NMR(苯-d6,25℃,300MHz):δ6.90(d,6H,3JHH=7.2Hz,間位-Ar-H),6.73(t,3H,3JHH=7.3Hz,對位-Ar-H)3.59(q,1H,3JHH=7.7Hz,Pn-H)2.28(s,18H,Ar-CH3)2.21,2.14,1.96,1.58,1.53(s,每3H,Pn-CH3)1.08(d,3H,3JHH=7.32Hz,Pn-CH3)。13C{1H}NMR(苯-d6,25℃,75.1MHz)δ164.05(本位-Ar),147.30,144.11,138.88,130.71(Pn),129.06(Ar),128.93(Pn),127.41(Ar),122.49(Pn),120.79(Ar),116.96(Pn),44.46(sp3Pn),18.15(sp3Pn-CH3),15.33,13.37,12.49,11.99,11.75,11.58(Pn-CH3)。
結晶學細節
[η8-(Pn*)Ti(O-2,6-Me-C6H3)Cl]單晶在-35℃下從甲苯溶液生長,C22H27ClOTi,Mr=390.81,三斜晶系,P-1,α=77.0104(7)°,β=89.3195(7)°,γ=85.2957(8)°,Z=4,T=150K,稜晶,紅棕色,9129獨立反射,R(初始)=0.038,R1=0.046wR2=0.133[I>2σ(I)]。
[η8-(Pn*)Ti(O-2,4-tBu-C6H3)Cl]單晶在-35℃下從苯溶液生長,C28H39ClOTi,Mr=474.97,單斜晶系,P21/n,α=102.4652(5)°,β=90°,γ=90°,Z=4,T=150K,塊狀,紫色,5884獨立反射,R(初始)=0.029,R1=0.038wR2=0.091[I>2σ(I)]。
[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]單晶在-35℃下從甲苯溶液生長,C30H36O2Ti,Mr=476.51,正交晶系,Pbca,α=90°,β=90°,γ=90(8)°,Z=8,T=150K,塊狀,深紅色,5807獨立反射,R(初始)=0.036,R1=0.047wR2=0.092[I>2σ(I)]。
[η8-(Pn*)Ti(OtBu)Cl]單晶在-35℃下從己烷溶液生長,C18H27ClOTi,Mr=342.76,三斜晶系,P-1,α=79.8812(7)°,β=78.4070(7)°,γ=73.9368(7)°,Z=2,T-150K,塊狀,深紅色,4073獨立反射,R(初始)=0.016,R1=0.034wR2=0.085[I>2σ(I)]。
[η8-(Pn*)Ti(OtBu)2]單晶在-35℃下從苯溶液生長,C22H36O2Ti,Mr=380.43,單斜晶系,P21/n,α=90°,β=93.2853(6)°,γ=90°,Z=4,T=150K,稜晶,紅棕色,5062獨立反射,R(初始)=0.022,R1=0.075wR2=0.188[I>2σ(I)]。
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)Cl2]單晶在-35℃下從己烷溶液生長,C22H28Cl2TiO,M=427.27,單斜晶系,P21/c,α=γ=90.00°,β=106.6445(11),T=150(2)K,Z=4,4844獨立反射,R(初始)=0.019,R1=0.054wR2=0.119[I>2σ(I)]。
[η5-(Pn*H)Ti(O-2,6-Me-C6H3)3]單晶在-35℃下從己烷溶液生長,C38H46TiO3,M=598.68,單斜晶系,P21/n,α=γ=90.00°,β=91.352(2),T=150(2)K,Z=4,6634獨立反射,R(初始)=0.060,R1=0.056wR2=0.141[I>2σ(I)]。
實驗細節II-涉及(氫)全甲基並環戊二烯
一般程序
所有有機金屬合成都在氮氣的惰性氣氛下利用標準Schlenk技術在雙重真空-進口氣體歧管或Braun手套箱上進行。在必要時,溶劑通過經以下合適的乾燥劑回流來乾燥:NaK(Et2O)、鈉(THF)以及SPS乾燥體系(己烷、戊烷、甲苯)。將溶劑在氮氣的流動的流下從乾燥劑蒸餾並且使用虹吸技術通過鋼插管來轉移並且在氮氣的氣氛下儲存在火焰乾燥的玻璃安瓿中。氘代的NMR溶劑在使用之前,經NaK(苯-d6、甲苯-d8)或CaH2(吡啶-d5)乾燥、真空轉移並且冷凍-抽吸-解凍-脫氣三次。元素分析由Stephen Boyer先生在倫敦都市大學的元素分析服務部進行。NMR光譜使用楊氏分接頭NMR管被記錄在Varian Mercury VX-Works 300MHz光譜儀上。1H光譜和13C{1H}NMR光譜以殘餘的質子溶劑峰作參考。
X射線結晶學
使用全氟聚醚油將晶體固定在玻璃纖維上,轉移至在衍射計上的測角計頭並且在冷的氮氣的流中使用Oxford Cryosystems CRYOSTREAM單元迅速冷卻至150K。數據收集使用Enraf-Nonius FR590KappaCCD衍射計利用石墨-單色化的Mo KαX射線輻射來進行。強度數據使用DENZO-SMN包來處理。結構使用直接法程序SIR92來求解,並且使用CRYSTALS程序組對所有F2數據使用全矩陣最小二乘法求精來求精。
聚合程序
將丙交酯單體(40mg)和絡合物遵循期望的單體:引發劑的比率引入NMR管中。然後,將0.57mL的氯仿-d1添加至化合物,產生[LA]0=0.5M的初始單體濃度。溶液通過1H NMR光譜學來監測。轉化率通過將聚合物相對於單體的甲烷面積積分來確定。
實施例8
Pn*(H)SnMe3的合成。在-78℃下向Pn*(H)Li(2.09g,10.7mmol)在戊烷(20mL)中的漿料添加SnMe3Cl(2.14g,10.7mmol)在戊烷(10mL)中的溶液。將反應混合物加溫至室溫並且攪拌持續3小時以提供橙色溶液和LiCl的無色沉澱。將此過濾並且將揮發物在真空中除去以提供作為橙色油的Pn*(H)SnMe3(通過1H NMR光譜學判斷的非對映異構體的50:50混合物)。收率:3.56g(97%)。1H NMR(苯-d6,23℃):δ2.98(q,1H,3JHH=7.2Hz,Pn*(H)),2.09 2.05 2.00(s,每3H,CH3-Pn*(H)),1.95(重疊的單峰,每3H,CH3-Pn*(H))1.93 1.83(s,每3H,CH3-Pn*(H)),1.70(重疊的單峰,每3H,CH3-Pn*(H)),1.59(s,3H,CH3-Pn*(H)),1.18(d,3H 3JHH=7.2Hz,1-CH3-Pn*(H)),0.94(d,3H,3JHH=6.9Hz,1-CH3-Pn*(H)),-0.01(s,9H,2J1H-119Sn=25.2Hz,2J1H-117Sn=24.2Hz,5-SnMe3-Pn*(H))-0.03(s,9H,2J1H-119Sn=25.3Hz,2J1H-117Sn=24.3Hz,5-SnMe3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):150.2 147.2 144.6 144.4 129.8 129.4 121.3 119.6(6x重疊的共振,(q-Pn*(H)),44.3 41.8(1-Pn*(H)),13.5 13.4 12.8 12.4 121.3 12.2 12.1 12.0(2x重疊的共振CH3-Pn*(H)),18.2 17.6(1-CH3-Pn*(H)),-8.8(5-SnMe3-Pn*(H),2J1H-119Sn=153Hz,2J1H-117Sn=148Hz),-9.2(5-SnMe3-Pn*(H),2J1H-119Sn=157Hz,2J1H-117Sn=150Hz)。
實施例9
Pn*(H)TiCl3的合成。向TiCl4(thf)2(0.408g,1.44mmol)在苯(2mL)中的漿料添加Pn*(H)SnMe3(0.505g,1.44mmol)在苯(2mL)中的溶液以提供深紫色溶液。將反應混合物加熱至80℃持續4小時。將揮發物在真空中除去以提供作為紫色粉末的Pn*(H)TiCl3。收率:0.363g(74%)。適於X射線衍射研究的單晶在-35℃下從飽和的Et2O溶液生長。1H NMR(苯-d6,23℃):δ0.85(d,3H,3JHH=7.5Hz,1-CH3-Pn*(H)),1.57 1.89 2.02 2.03 2.14(s,每3H,CH3-Pn*(H)),3.80(q,1H,3JHH=8.5Hz,Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ153.6 152.7 147.4 140.6 133.3 131.3 127.4(q-Pn*(H)),46.8(1-Pn*(H)),15.4(1-CH3-Pn*(H)),14.5,14.4,14.2,12.2,11.6(CH3-Pn*(H))。
實施例10
[Pn*(H)ZrCl3]2的合成。向ZrCl4(0.995g,4.27mmol)在苯(5mL)中的漿料添加Pn*(H)SnMe3(1.50g,4.27mmol)在苯(5mL)中的溶液。將反應混合物加熱至80℃持續72小時以提供深綠色溶液。將揮發物在真空中除去以產生綠色固體。向此添加戊烷(15mL)並且將反應混合物聲處理持續15分鐘以提供細的橄欖綠色的粉末和淺黃色溶液。將反應混合物過濾並且將濾液在減壓下乾燥以提供作為橄欖綠色粉末的[Pn*(H)ZrCl3]2。收率:1.42g(87%)。單晶在23℃下從飽和的苯溶液生長。1H NMR(苯-d6,23℃):δ0.92(d,3H,3JHH=7.5Hz,1-CH3-Pn*(H)),1.81 2.01 2.06 2.17 2.19(s,每3H,CH3-Pn*(H)),3.50(q,3JHH=7.5Hz,Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ151.0 147.0 142.1 133.5 129.5 127.4 120.7(q-Pn*(H),46.0(1-Pn*(H)),15.6(1-CH3-Pn*(H)),14.2 13.6 13.5 12.3 12.2(CH3-Pn*(H))。
實施例11
[Pn*(H)HfCl3]2的合成。向HfCl4(0.164g,0.467mmol)在苯(2mL)中的漿料添加Pn*(H)SnMe3(0.149g,0.467mmol)在苯(2mL)中的溶液。將反應混合物加熱至80℃持續2小時以提供橙色溶液。將揮發物在真空中除去以產生作為淺黃色固體的[Pn*(H)HfCl3]2。單晶在室溫下從飽和的苯溶液生長。1H NMR(苯-d6,23℃):δ0.93(d,3H,3JHH=7.4Hz,1-CH3-Pn*(H)),1.621.84 2.01 2.01 2.10(s,每3H,CH3-Pn*(H)),3.42(q,3JHH=7.4Hz,Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ150.0 142.3 137.2 123.9 117.4(q-Pn*(H)),45.5(1-Pn*(H)),15.9(1-CH3-Pn*(H)),12.2,12.1,12.0,11.9,11.6(CH3-Pn*(H))。
實施例12
η5-Pn*(H)Ti(OtBu)3的合成。將Pn*(H)TiCl3(0.020g,0.059mmol)和KOtBu(0.020g,0.18mmol)在苯-d6(0.5mL)中合併並且聲處理持續5分鐘以提供澄清的淺黃色溶液和無色沉澱。過濾,隨後在真空中乾燥濾液,提供作為淺黃色粉末的Pn*(H)Ti(OtBu)3,1。1H NMR(苯-d6,23℃):δ3.35(q,3JHH=7.2Hz,Pn*(H)),2.27 2.24 2.12 2.08 1.84(s,每3H,CH3-Pn*(H)),1.30(s,27H,OC(CH3)3),1.25(d,3H 3JHH=7.3Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ141.5 135.1 132.9 128.6 121.0 115.6 110.5(q-Pn*(H)),75.4(OC(CH3)3),43.7(1-Pn*(H)),33.4(OC(CH3)3),16.2(1-CH3-Pn*(H)),12.712.6 12.1 12.1 11.4(CH3-Pn*(H))。
實施例13
η5-Pn*(H)Zr(OtBu)3的合成。將[Pn*(H)ZrCl3]2(0.028g,0.036mmol)和KOtBu(0.024g,0.22mmol)在苯-d6(0.5mL)中合併並且聲處理持續5分鐘以提供澄清的淺黃色溶液和無色沉澱。過濾,隨後在真空中乾燥濾液,提供作為淺黃色粉末的Pn*(H)Zr(OtBu)3。1H NMR(苯-d6,23℃):δ3.33(q,3JHH=6.9Hz,Pn*(H)),2.25 2.22 2.09 2.05 1.83(s,每3H,CH3-Pn*(H)),1.35(s,27H,OC(CH3)3),1.23(d,3H,3JHH=6.9Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ142.8 137.5 134.4 129.3 123.4 117.8 112.9(q-Pn*(H)),80.2(OC(CH3)3),43.8(1-Pn*(H)),33.1(OC(CH3)3),15.9(1-CH3-Pn*(H)),13.7,12.8,12.7,12.3 12.2(CH3-Pn*(H))。
實施例14
η5-Pn*(H)Zr(O-CH2C6H5)3的合成。將[Pn*(H)ZrCl3]2(0.100g,0.131mmol)和KO-CH2C6H5(0.115g,0.786mmol)在C6H6(5mL)中合併並且攪拌持續10分鐘以提供澄清的淺黃色溶液和無色沉澱。過濾,隨後在真空中乾燥濾液,在室溫下提供作為淺黃色油狀固體的Pn*(H)Zr(O-CH2C6H5)3。1H NMR(苯-d6,23℃);δ7.37–7.03(重疊的多重峰,15H,CH2C6H5),5.10(s,6H,CH2C6H5),3.12(q,1H,3JHH=7.3Hz,Pn*(H)),2.13 2.09 1.99 1.90 1.68(s,每3H CH3-Pn*(H)),1.13(d,3H 3JHH=7.3Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ143.9(CH2-1-C6H5),143.0 135.4 133.2(q-Pn*(H)),128.5(CH2-2,3,4-C6H5),127.1(q-Pn*(H)),126.9 126.4(CH2-2,3,4-C6H5),122.0 116.5 111.3(q-Pn*(H)),71.7(CH2C6H5),43.2(1-Pn*(H)),16.2(1-CH3-Pn*(H)),12.3 11.8 11.5 11.0 10.4(CH3-Pn*(H))。
實施例15
η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3的合成。將Pn*(H)ZrCl3和S-KOCH{CH3}C6H5在苯(5mL)中合併並且攪拌持續10分鐘以提供澄清的淺黃色溶液和無色沉澱。過濾,隨後在真空中乾燥濾液,在室溫下提供作為淺黃色油狀固體的η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3。1H NMR(苯-d6,23℃):兩種非對映異構體:δ7.40(d,6H,3JHH=7.3Hz,2,6-C6H5),7.11(m,3H,4-C6H5),5.30(q,3H,3JHH=6.1Hz,CHMe),3.14 3.09(q,3JHH=6.9Hz,Pn*(H)),2.11 2.11 2.08 2.05 1.97 1.97 1.89 1.88 1.70 1.65(重疊的單峰,每3H,CH3-Pn*(H)),1.44(d,9H,3JHH=6.1Hz,CHMe),1.12(d,3H,3JHH=6.9Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):非對映異構體1:δ148.7142.7 135.5 133.3(q-Pn*(H)),128.6(3,5-C6H5),126.9(2,6-C6H5),125.7(q-Pn*(H)),125.7(4-C6H5),122.0 116.4 111.4(q-Pn*(H)),77.0(CHMe),43.4(1-Pn*(H)),28.4(CHMe),16.1(1-CH3-Pn*(H)),12.2 11.8 11.8 11.2 10.6(CH3-Pn*(H))。非對映異構體2:δ148.7 142.7 135.3 133.2(q-Pn*(H)),128.5(3,5-C6H5),126.9(2,6-C6H5),125.9(4-C6H5),125.5(q-Pn*(H)),121.8 116.4 111.1(q-Pn*(H)),77.0(CHMe),43.3(1-Pn*(H)),28.4(CHMe),16.1(1-CH3-Pn*(H)),12.2 11.8 11.7 11.2 10.5(CH3-Pn*(H))。
實施例16
η5-Pn*(H)Zr(O-外消旋-{CH3}C6H5)3的合成。將Pn*(H)ZrCl3和外消旋-KOCH{CH3}C6H5在苯(5mL)中合併並且攪拌持續10分鐘以提供澄清的淺黃色溶液和無色沉澱。過濾,隨後在真空中乾燥濾液,在室溫下提供作為淺黃色油狀固體的η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3。1H NMR(苯-d6,23℃):非對映異構體的混合物:δ7.48-7.10(重疊的多重峰,15H,C6H5),5.24(重疊的四重峰,3H,CHMe),3.06(q,1H,3JHH=7.3Hz,Pn&(H)),2.12 2.12 2.08 2.07 1.98 1.98 1.90 1.69 1.67(重疊的單峰,每3H,CH3-Pn*(H)),1.50 1.45 1.44 1.41(重疊的雙峰,9H 3JHH=6.4Hz,CHMe),1.12(d,3H,3JHH=7.3Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):非對映異構體的混合物:δ148.7 142.7 135.5 133.3(q-Pn*(H)),128.4(3,5-C6H5),127.3(q-Pn*(H)),126.9(2,6-C6H5),125.6(4-C6H5),122.0 116.4 111.4(q-Pn*(H)),77.0(CHMe),43.3(1-Pn*(H)),28.4(CHMe),16.1(1-CH3-Pn*(H)),12.2 11.8 11.7 11.2 10.6(CH3-Pn*(H))。在13C{1H}NMR光譜中報導的值是觀察到的多重重疊的共振的中心值。
實施例17
η5-Pn*(H)Zr(O-2,6-Me-C6H3)3的合成。將[Pn*(H)ZrCl3]2和KO-2,6-Me2C6H3在苯(5mL)中合併並且攪拌持續10分鐘以提供澄清的淺黃色溶液和無色沉澱。過濾,隨後在真空中乾燥濾液,提供作為淺黃色粉末的Pn*(H)Zr(O-2,6-Me-C6H3)3。1H NMR(苯-d6,23℃):δ6.93(d,6H,3JHH=7.4Hz,3,5-C6H3),6.75(t,3H,3JHH=7.3Hz,4-C6H3,3.31(q,3JHH=7.4Hz,Pn*(H)),2.25(s,18H,O-2,6-CH3-C6H3),2.20 2.12 1.96 1.69 1.54(s,每3H,CH3-Pn*(H)),1.09(d,3H,3JHH=7.4Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ159.7(1-C6H3),145.3 139.7 135.6(q-Pn*(H)),128.9(3,5-C6H3),128.0(q-Pn*(H)),126.6(2,6-C6H3),125.7(q-Pn*(H)),120.4(4-C6H3),119.0 113.7(q-Pn*(H)),43.9(q-Pn*(H)),17.9(O-2,6-CH3-C6H3),15.7(1-CH3-Pn*(H)),12.5 11.8 11.7 11.0(CH3-Pn*(H))。
實施例18
η5-Pn*(H)Zr(O-2,6-iPr-C6H3)3的合成。將[Pn*(H)ZrCl3]2(0.174g,0.225mmol)和KO-2,6-iPr2C6H3(0.2925g,1.35mmol)在苯(5mL)中合併並且攪拌持續10分鐘以提供澄清的淺黃色溶液和無色沉澱。過濾,隨後在真空中乾燥濾液,提供作為黃綠色粉末的η5-Pn*(H)Zr(O-2,6iPr-C6H3)3。對於C50H70O3Zr的分析計算值:C,74.11;H,8.71。實測值:C,67.68;H,8.60。1H NMR(苯-d6,23℃):δ7.11-7.03(重疊的雙峰,6H,3,5-C6H3),6.96(明顯的三重峰,3H,3JHH=6.7Hz,4-C6H3),3.52(重疊的七重峰,6H 3JHH=6.7Hz,CH(CH3)2),3.11(q,1H,3JHH=7.4Hz,Pn*(H)),2.17 2.13 1.93 1.85 1.44(s,每3H,CH3-Pn*(H)),1.32(d,12H,3JHH=6.7Hz,CH(CH3)2,1.01(d,3H,3JHH=7.4Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ155.9 155.8(1-C6H3),146.8(q-Pn*(H)),140.6(q-Pn*(H)),137.9 137.7(2,6-C6H3),136.4 127.4(q-Pn*(H)),123.6 123.5(3,5-C6H3),122.1 122.1(4-C6H3)120.5 116.2(q-Pn*(H)),43.3(1-Pn*(H)),27.3 27.2(CH(CH3)2),24.9 24.8 24.0 23.9(CH(CH3)2),15.6(1-CH3-Pn*(H)),12.1 12.0 12.0 11.8 10.9(CH3-Pn*(H))。對Pn*(H)碳作出解釋的一個四級共振與殘餘的質子溶劑共振重疊。
實施例19
η5-Pn*(H)ZrCl2(O-2,6-tBu-C6H3)的合成。將[Pn*(H)ZrCl3]2(0.239g,0.310mmol)和KO-2,6-tBu2C6H3(0.152g,0.62mmol)在苯(5mL)中合併並且攪拌持續10分鐘以提供澄清的淺黃色溶液和無色沉澱。過濾,隨後在真空中乾燥濾液,提供作為黃綠色粉末的η5-Pn*(H)ZrCl2(O-2,6-tBu-C6H3)。收率:0.072g(42%)。1H NMR(苯-d6,23℃):δ7.13(d,2H,3JHH=7.7Hz,3,5-C6H3),6.78(t,1H,3JHH=7.7Hz,4-C6H3),2.96(q,1H,3JHH=7.4Hz,Pn*(H)),2.20 2.05 1.98 1.69(s,每3H,CH3-Pn*(H)),1.41(寬的單峰,18H,C(CH3)3),1.32(s,3H,CH3-Pn*(H)),0.86(d,6H,3JHH=7.4Hz,CH(CH3)2)。13C{1H}NMR(苯-d6,23℃):δ161.0(1-C6H3),148.2 145.9 134.4 132.3(q-Pn*(H)),125.4(3,5-C6H3),125.2(q-Pn*(H)),121.4(4-C6H3),119.6(q-Pn*(H)),44.7(1-Pn*(H)),35.6(C(CH3)3),32.1(C(CH3)3),15.5(1-CH3-Pn*(H)),14.0 12.9 12.4 12.4 116(CH3-Pn*(H))。對2,6-C6H3和Pn*(H)碳作出解釋的兩個四級共振與殘餘的質子溶劑共振重疊。
實施例20
η5-Pn*(H)Hf(O-2,6-Me-C6H3)3的合成。將[Pn*(H)HfCl3]2和KO-2,6-Me2C6H3在苯(5mL)中合併並且攪拌持續10分鐘以提供澄清的淺黃色溶液和無色沉澱。過濾,隨後在真空中乾燥濾液,提供作為淺黃色粉末的Pn*(H)Hf(O-2,6-Me-C6H3)3。1H NMR(苯-d6,23℃):δ6.93(d,6H,3JHH=7.4Hz,3,5-C6H3),6.74(t,3H,3JHH=7.4Hz,4-C6H3),3.27(q,1H,3JHH=7.4Hz,4-C6H3),3.27(q,1H,3JHH=7.4Hz,Pn*(H)),2.24(s,18H,O-2,6-CH3-C6H3),2.24 2.16 1.99 1.68 1.56(s,每3H,CH3-Pn*(H)),1.10(d,3H,3JHH=7.4Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ159.4(1-C6H3),145.1 138.7 134.0(q-Pn*(H)),129.0(3,5-C6H3),127.8(q-Pn*(H)),126.9(2,6-C6H3),124.1(q-Pn*(H)),120.4(4-C6H3),117.6 112.1(q-Pn*(H)),43.9(1-Pn*(H)),17.8(O-2,6-CH3-C6H3),15.5(1-CH3-Pn*(H)),12.4 11.8 11.6 11.4 10.9(CH3-Pn*(H))。
實施例21
η5-Pn*(H)HfCl(O-2,6-iPr-C6H3)2的合成。將[Pn*(H)HfCl3]2和KO-2,6-iPr2C6H3在苯(5mL)中合併並且攪拌持續10分鐘以提供澄清的淺黃色溶液和無色沉澱。過濾,隨後在真空中乾燥濾液,提供作為淺黃色粉末的η5-Pn*(H)HfCl(O-2,6iPr-C6H3)2。1H NMR(苯-d6,23℃):δ7.10-6.86(重疊的多重峰,6H,3,4,5-C6H3),3.52(重疊的七重峰,4H,CH(CH3)2),3.11(q,1H,3JHH=7.4Hz,Pn*(H)),2.25 2.20 1.92 1.92 1.49(s,每3H,CH3-Pn*(H)),1.30(d,12H,3JHH=6.5Hz,CH(CH3)2),1.24 1.23(重疊的雙峰,每6H,3JHH=6.5Hz,CH(CH3)2)1.05(d,3H,3JHH=7.4Hz,1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ155.7 155.6(1-C6H3),146.3 139.1(q-Pn*(H)),137.9 137.8(2,6-C6H3),134.7 127.6 125.6(q-Pn*(H)),123.9 123.5(3,5-C6H3),122.1 122.0(4-C6H3),118.5 114.5(q-Pn*(H)),43.5(1-Pn*(H)),27.1 27.0(CH(CH3)2),24.9 24.9 24.1 24.0(CH(CH3)2),15.5(1-CH3-Pn*(H)),11.9 11.8 10.8(CH3-Pn*(H))。
實施例22
η5-Pn*(H)HfCl2(O-2,6-tBu-C6H3)的合成。將[Pn*(H)HfCl3]2和KO-2,6-tBu-C6H3在苯(5mL)中合併並且攪拌持續10分鐘以提供澄清的淺黃色溶液和無色沉澱。η5-Pn*(H)HfCl2(O-2,6-tBu-C6H3)通過NMR光譜學被鑑定為唯一的產物。NMR收率:(99.5%)。1H NMR(苯-d6,23℃):δ7.19(d,2H,3JHH=7.7Hz,3,5-C6H3),6.80(t,1H,3JHH=7.7Hz,4-C6H3),2.95(q,1H,3JHH=7.4Hz,Pn*(H)),2.29 2.16 2.06 1.71(s,每3H,CH3-Pn*(H)),1.42和1.44(寬的重疊的單峰,18H,C(CH3)3),1.36(s,3H,CH3-Pn*(H)),0.91(d,3H,3JHH=7.4Hz 1-CH3-Pn*(H))。13C{1H}NMR(苯-d6,23℃):δ160.7(1-C6H3),147.5 143.3 133.0 129.6 127.6(q-Pn*(H)或2,6-C6H3),125.4(3,5-C6H3),123.0(q-Pn*(H)或2,6-C6H3),121.2(4-C6H3),117.7(q-Pn*(H)或2,6-C6H3),44.5(1-Pn*(H)),35.6(C(CH3)3),32.1(C(CH3)3,15.5(1-CH3-Pn*(H)),13.7,12.6 12.0 11.4(CH3-Pn*(H))。對2,6-C6H3或Pn*(H)碳作出解釋的一個四級共振與殘餘的溶劑共振重疊。
實驗細節III-涉及基於茚基的催化劑
一般細節
空氣和水分敏感的化合物在氮氣的惰性氣氛下使用標準Schlenk管線技術在雙重歧管真空/氮氣管線或Braun Unilab手套箱上來操作。反應溶劑(戊烷、己烷、以及甲苯)使用MBraun SPS-800溶劑純化系統來乾燥。將己烷、甲苯、以及戊烷儲存在預活化的分子篩上。將乾燥的溶劑在氮氣的氣氛下儲存在烘箱乾燥的安瓿中,用Rotoflo或楊氏分接頭密封。將在空氣敏感的化合物的NMR分析中使用的氘代的溶劑在使用之前經合適的乾燥劑乾燥、冷凍-解凍脫氣並且真空轉移:將氯仿-d1(Sigma-Aldrich)儲存在預活化的分子篩上。NMR光譜被記錄在300MHz Varian Mercury VX-Works光譜儀上。除非另有陳述,否則1H(300.27MHz)光譜和13C{1H}(75.50MHz)光譜在25℃下被記錄,並且在內部以在使用的氘代溶劑中的殘餘的質子溶劑峰作為參考。1H化學位移和13C{1H}化學位移δ以百萬分率(ppm)被給出,相對於殘餘的溶劑峰被給出。空氣敏感的樣品在惰性氣氛中在手套箱中使用在楊氏分接頭NMR管中乾燥的溶劑來製備。[(Ind)2ZrMe2]和絡合物根據文獻程序被合成。將[(EBI)ZrCl2](Strem Chemicals)在甲苯中熱重結晶(hot re-crystallise)。
聚合程序
所有聚合都在包含40mg的在引發劑的氯仿-d1溶液(20mg的在4mL氯仿-d1中的引發劑)中的丙交酯的楊氏分接頭NMR管中進行,確保丙交酯:引發劑的比率是50:1。然後,添加氯仿-d1以確保初始的丙交酯濃度是[LA]0=0.50M。包括添加叔丁醇的聚合照常被製備,並且將叔丁醇經由微注射器添加至氯仿-d1溶液,確保了丙交酯:引發劑:叔丁醇的比率是50:1:1。
X射線結晶學
使用全氟聚醚油將晶體固定在MiTeGen MicroMants上,並且在冷的氮氣的流中使用Oxford Cryosystems CRYOSTREAM單元迅速冷卻至150K。數據收集使用Enraf-Nonius FR590KappaCCD衍射計、利用石墨-單色化的Mo KαX射線輻射來進行。原始幀數據(Raw frame data)在150(2)K下使用Nonius KappaCCD衍射計收集,使用DENZO-SMN歸納並且使用SORTAV對吸收作出校正。使用SuperFlip解出結構並且使用CRYSTALS用全矩陣最小二乘法求精。
[EBI)Zr(O-2,6-Me-C6H3)Cl]的結晶學數據
單晶在室溫下從甲苯溶液生長,C28H25BClOZr,Mr=504.17,正交晶系,Pcab,α=90°,β=90°,γ=90°,Z=6,T=150K,塊狀,黃色,10201獨立反射,R(初始)=0.070,R1=0.057wR2=0.137[I>2σ(I)]。
實施例23
[EBI)Zr(O-2,6-Me-C6H3)Cl]的合成
在室溫下向一當量的在甲苯(10mL)中的[EBI)ZrCl2](100mg,0.24mmol)添加一當量的在甲苯(10mL)中的2,6-二甲基苯酚(29mg,0.24mmol)。將黃色懸浮液留下攪拌持續18小時,產生澄清的黃色溶液。將溶劑在真空中除去,以提供作為黃色結晶固體的[(EBI)Zr(O-2,6-Me-C6H3)Cl],收率為80%(96mg,0.19mmol)。1H NMR(氯仿-d1,25℃,300MHz):δ7.84(1H,dd,ArH,3JHH=8.7Hz,4JHH=0.9Hz),7.61(1H,dd,ArH,3JHH=8.6Hz,4JHH=0.9Hz),7.30–7.27(2H,m,ArH),7.20–7.10(2H,m,ArH),7.03(1H,t,ArMe2H,3JHH=7.5Hz),6.78(2H,d,ArMe2H,3JHH=7.4Hz),6.63–6.55(2H,m,ArH),6.54(1H,d,CpH,3JHH=3.2Hz),6.41(1H,d,CpH,3JHH=3.2Hz),6.15(1H,d,CpH,3JHH=3.2Hz),6.02(1H,d,CpH,3JHH=3.2Hz),3.88-3.62(4H,m,橋),1.91(6H,s,ArMe2)。13C{1H}NMR(氯仿-d1,25℃,75.5Mhz):δ128.2,126.6,125.7 125.6,125.4,123.7,123.6,122.7 121.7,120.9,119.5,115.7,113.2,108.4,106.1(全部非四級環碳)29.6(C2H4),28.7(C2H4),18.0(2xArMe2)。季碳未賦值。
實施例24
[Ind2Zr(O-2,6-Me-C6H3)Me]的合成
在室溫下向一當量的在甲苯(5mL)中的[Ind2ZrMe2](100mg,0.28mmol)添加一當量的在甲苯(5mL)中的2,6-二甲基苯酚(34mg,0.28mmol)。將澄清的稻草色溶液留下攪拌持續18小時。將溶劑在真空中除去,以提供作為無色油的[(Ind)2Zr(O-2,6-Me-C6H3)Me],收率為75%(0.21mmol,96mg)。1H NMR(氯仿-d1,25℃,300Mhz):δ7.48-7.43(2H,m,ArH),7.34-7.29(2H,m,ArH),7.07-7.00(2H,m,ArH),6.92-6.85(2H,m,ArH),6.82(2H,d,ArMe2H,3JHH=7.4Hz),6.59(1H,t,ArMe2H,3JHH=7.4Hz),6.20(2H,m,CpH),5.91(2H,m,CpH),5.76(2H,t,CpH,3JHH=3.4Hz),1.86(6H,s,ArMe2),0.22(3H,s,ZrMe)。13C{1H}NMR(氯仿-d1,25℃,75.5Mhz):δ159.4(CO,Ar),128.0(2x CH,Ar),125.7(2x C四級,Ar),124.6(2x CH,Ar),124.3(2x CH,Ar),124.0(2x C四級,Ar),123.9(2x CH,Ar),123.8(2x C四級,Ar),123.6(2x CH,Ar),118.9(CH,Ar),117.6(2x CH,Cp),100.6(2x CH,Cp),99.0(2x CH,Cp),27.8(ZrMe),17.6(2x ArMe2)。
實施例-丙交酯的聚合
(I)
L-丙交酯和外消旋-丙交酯的聚合
L-丙交酯單體和外消旋-丙交酯單體
為了研究芳氧基(aryloxide group)之間的差異,以50的單體:引發劑的比率在100℃下在氯仿-d1中進行丙交酯單體的聚合的偽一級動力學數據(pseudo-first order kinetic data)。結果在表1中被示出並且在圖1中被圖示。觀察到的增長速率k觀察通過分析1n([LA]0/[LA]t)相對於時間的半對數標繪圖來確定,其中[LA]0=0.50mol/L。
表1. L-丙交酯聚合:芳氧基取代基的變化
聚合條件:100℃,[LA]0=50,[LA]0=0.5M,氯仿-d1。
圖1圖示使用以下絡合物的L-丙交酯聚合:[η5-Pn*(H)Ti(O-2,6-Me-C6H3)3 2(黑色正方形,k觀察=0.113±0.014h-1)、η5-Pn*(H)Zr(O-2,6-Me-C6H3)3 7(紅色圓形,k觀察=0.479±0.032h-1)、η5-Pn*(H)Zr(O-2,6-iPr-C6H3)3 8(粉色三角形,k觀察=0.391±0.022h-1)、η5-Pn*(H)ZrCl2(O-2,6-tBu-C6H3)9(深藍色三角形,k觀察=0.043±0.003h-1)、η5-Pn*(H)Hf(O-2,6-Me-C6H3)3 10(藍色三角形,k觀察=0.364±0.027h-1)、η5-Pn*(H)HfCl(O-2,6iPr-C6H3)2 11(綠色菱形,k觀察=0.463±0.029h-1)以及η5-Pn*(H)HfCl2-2,6tBu-C6H3)12(紫色三角形,k觀察=0.086±0.020h-1)。聚合條件:氯仿-d1,在100℃下,其中[LA]0/[M]0=50,[LA]0=0.5M。
如在圖1中可以看到,基於二甲基和二異丙基芳族基團與鋯和鉿的四種絡合物(7、8、10以及11)產生最高的增長常數(0.364<k觀察<0.479h-1)。兩種叔丁基絡合物(9和12)均證明最低的聚合速率(對於η5-Pn*(H)ZrCl2(O-2,6-tBu-C6H3)具有0.043h-1的k觀察,而對於η5-Pn*(H)HfCl2(O-2,6-tBu-C6H3)具有0.086h-1的k觀察))。當使用二甲基酚鹽類型絡合物改變金屬時,出現的是,鋯比鉿比鈦更迅速(分別地,0.479h-1、0.391h-1以及0.113h-1的k觀察)。這些速率在文獻的範圍內。
當然由於聚合的高溫度,多分散性是相對地高的(1.45<Mw/Mn<1.74)。分子量比理論的分子量略微較高(7,547g/mol<Mn<11,680g/mol)。
烷氧基取代基(alkoxide group substituent)的變化對L-丙交酯的聚合的影響已經在100℃下以50的單體:引發劑的比率在氯仿-d1中被研究。當使用絡合物η5-Pn*(H)Ti(OtBu)3 1和η5-Pn*(H)Zr(OtBu)3 3時,在1小時之後未實現轉化。然而,將叔丁基改變為苄基型取代基顯著地增大了聚合的速率,對於η5-Pn*(H)Ti(O-CH2Ph)3 4、η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3 5、η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3 6,具有高於90%的轉化率(分別地,93%、96%以及97%)。
為了研究手性對引發基團的影響,以50的單體:引發劑的比率在100℃下在氯仿-d1中進行丙交酯單體的聚合的偽一級動力學數據。結果在表2中被示出並且在圖2-3和SI中被圖示。
聚合條件:[LA]0/[M]0=50,[LA]0=0.5M,氯仿-d1。
圖2示出丙交酯聚合:L-丙交酯和η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3,5(黑色正方形,k觀察=1.166±0.068h-1);L-丙交酯和η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3,6(紅色圓形,k觀察=1.954±0.063h-1);外消旋-丙交酯和η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3,6(粉色三角形,k觀察=1.667±0.053h-1);外消旋-丙交酯和η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3,5(藍色三角形,k觀察=1.342±0.055h-1)。聚合條件:氯仿-d1,在80℃下,其中[LA]0/[M]0=50,[LA]0=0.5M。
圖3示出丙交酯聚合:L-丙交酯和η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3,5(黑色正方形,k觀察=0.484±0.037h-1);L-丙交酯和η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3,6(紅色圓形,k觀察=0.850±0.063h-1);外消旋-丙交酯和η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3,6(粉色三角形,k觀察=0.767±0.037h-1);外消旋-丙交酯和η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3,5(藍色三角形,k觀察=0.491±0.031h-1)。聚合條件:氯仿-d1,在60℃下,其中[LA]0/[M]0=50,[LA]0=0.5M。
兩種絡合物η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3 5、η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3 6對於L-丙交酯和外消旋-丙交酯在100℃下均證明非常高的聚合速率(1.885h-1<k觀察<3.442h-1)。在三個溫度(60℃、80℃以及100℃)下,對於兩種丙交酯單體,外消旋絡合物聚合得更迅速。在60℃下,當使用外消旋-η5-Pn*(H)Zr(O-CH{CH3}C6H5)3 6時觀察到的增長速率(對於L-丙交酯為0.850h-1的k觀察並且對於外消旋-丙交酯為0.767h-1的k觀察),比當使用η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3 5時(對於L-丙交酯為0.484h-1的k觀察並且對於外消旋-丙交酯為0.491h-1的k觀察)快約70%。L-丙交酯和外消旋-丙交酯似乎以類似的聚合速率被聚合。
多分散性隨降低的溫度而降低(對於100℃,1.27<Mw/Mn<1.44並且對於80℃,1.15<Mw/Mn<1.24)。分子量實驗值是理論值的一半(2,693g/mol<Mn<4,549g/mol)。
使用η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3 5、η5-Pn*(H)Zr(O-外消旋-CH{CH3}C6H5)3 6引發的用於L-丙交酯和外消旋-丙交酯的開環聚合的活化參數使用艾林標繪圖來確定並且被發現是30.4kJ/mol<ΔH#<46.6kJ/mol且411.4J/(mol K)<ΔS#<640J/(mol K),圖4和SI。
圖4是使用η5-Pn*(H)Zr(O-S-CH{CH3}C6H5)3 5的L-丙交酯聚合的艾林標繪圖。斜率=-5610±488,且R2=0.993。聚合條件:氯仿-d1,其中[LA]0/[Zr]0=50並且[LA]0=0.5M。
L-丙交酯的聚合的偽一級動力學數據用於使用η5-Pn*(H)Zr(O-2,6iPr-C6H3)3 8在100℃下在氯仿-d1中研究單體濃度:引發劑濃度的比率的變化的影響。結果在表3中被核對並且在圖5中被圖示。
表3. L-丙交酯聚合:濃度的變化
聚合條件:100℃,[LA]0=0.5M,氯仿-d1。
圖5示出使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8的L-丙交酯聚合:[LA]0/[Zr]0–25(黑色正方形,k觀察=0.521±0.021h-1);[LA]0/[Zr]0=50(紅色圓形,k觀察=0.391±0.022h-1);[LA]0/[Zr]0=100(藍色三角形,k觀察=0.377±0.015h-1);[LA]0/[Zr]0=200(粉色三角形,k觀察=0.235±0.004h-1)。聚合條件:氯仿-d1,在100℃下,其中[LA]0=0.5M。
如所預計,L-丙交酯在100℃下在氯仿-d1中的聚合速率隨著降低的單體:引發劑的比率而增大(分別地,對於200、100、50以及25的[LA]0/[Zr]0,0.235h-1、0.377h-1、0.391h-1以及0.521h-1的k觀察)。此外,分別地,對於25至200的初始單體:引發劑的比率,分子量Mn隨著增加的濃度從6,920g/mol增大至17,042g/mol。多分散性,Mw/Mn在1.40<Mw/Mn<1.74之間變化。
使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8的L-丙交酯的觀察到的增長速率證明了作為引發劑的函數的偽一級動力學(圖6)。
圖6示出使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8的Ln(k觀察)相對於Ln([ZR]0)的標繪圖。斜率=0.350±82,且R2=0.901。聚合條件:氯仿-d1,在100℃下並且[LA]0=0.5M。
L-丙交酯的聚合的偽一級動力學數據用於使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8在氯仿-d1中以50的單體:引發劑的比率研究溫度的影響。結果在表4中被核對並且在圖7中被圖示。
表4. L-丙交酯聚合:溫度的變化
聚合條件:[LA]0/[M]0=50,[LA]0=0.5M,氯仿-d1。
圖7示出使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8的L-丙交酯聚合:T=100℃(黑色正方形,k觀察=0.391±0.022h-1);T=90℃(紅色圓形,k觀察=0.151±0.008h-1);T=80℃(藍色三角形,k觀察-0.092±0.006h-1)。聚合條件:氯仿-d1,其中[LA]0/[M]0=50,[LA]0-0.5M。
如所預計,L-丙交酯在氯仿-d1中的聚合速率隨著降低的溫度而增大(分別地,對於100、90以及80的T,0.391h-1、0.151h-1以及0.092h-1的k觀察)。分子量Mn在6,348g/mol至7,751g/mol之間保持不變,這與理論的分子量非常接近。然而,多分散性Mw/Mn分別隨著從100℃至80℃的降低的溫度而從1.72降低至1.58。
用於使用η5-Pn*(H)Zr(O-2,6-Pr-C6H3)3 8引發的L-的開環聚合的活化參數使用艾林標繪圖來確定並且被發現是ΔH#=75.9kJ/mol和ΔS#=1847J/(mol K),圖8。
圖8示出使用η5-Pn*(H)Zr(O-2,6-Pr2C6H3)3 8的L-丙交酯聚合的艾林標繪圖。斜率=-9133±1881,且R2=0.959。聚合條件:氯仿-d1,其中[LA]0/[Zr]0=50並且[LA]0=0.5M。
合成的聚丙交酯通過1H、1H{1H}以及13C{1H}NMR光譜學來表徵。NMR光譜證明了當L-丙交酯被聚合時沒有差向異構並且當外消旋-丙交酯被聚合時等規立構偏向的PLA(an isotactic biased PLA)。
它們還已經通過MALDI-TOF和13C{1H}NMR光譜學來表徵以確定鏈的終止。示出的是,丙交酯單體插入在金屬-氧鍵中。
(II)
圖9中示出了在80℃下在苯-d6中、以1:50的引發劑:單體的比率、使用被選擇的全甲基並環戊二烯絡合物的L-丙交酯的聚合的偽一級動力學數據。用於所有聚合的動力學數據展現出取決於引發的絡合物而變化的誘導期。同樣地,在圖9中的數據從32小時被給出,直到那時所有誘導期已結束。
迄今為止展現出朝向丙交酯異構體的開環聚合的最高活性的絡合物是[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]。其證明了與公布的鈦絡合物(k觀察=69.9x10-3h-1)37類似並且在80℃下是絡合物[η8-(Pn*)Ti(O-2,6-Me-C6H3)Cl]和[η8-(Pn*)Ti(O-2,4-Bu-C6H3)Cl]的10倍的聚合速率,絡合物[η8-(Pn*)Ti(O-2,6-Me-C6H3)Cl]和[η8-(Pn*)Ti(O-2,4-Bu-C6H3)Cl]證明了類似的增長速率(分別地,k觀察=7.2x 10-3h-1和7.0x 10-3h-1)。這些增長速率是當使用[η5-(Pn*)Ti(O-2,6-Me-C6H3)Cl2](k觀察=1.9x 10-3h-1)時的3.5倍。
圖9示出L-丙交酯轉化率相對於時間的半對數標繪圖,[LA]0/[初始]0=50,[LA]0=0.104M,T=80℃,苯-d6(0.5mL),使用[η8-(Pn*)Ti(O-2,6-MeC6H3)Cl](綠色點劃線)、[η8-(Pn*)Ti(O-2,4-tBu-C6H3)Cl](黑色短劃線)、[η8-(Pn*)Ti(O-2,6-Me-C6H3)2](紅色線)以及[η5-(Pn*H)Ti(O-2,6-Me-C6H3)Cl2](藍色點虛線)。省略誘導期。
如在圖10中所示出的,在90℃下,當通過[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]引發時,外消旋-丙交酯和L-丙交酯以相同的速率(k觀察≈110x 10-3h-1)聚合。這可推測地是因為非手性引發劑不能在兩種丙交酯對映異構體之間區分並且已經以相同的速率併入每個。此立體化學優先性的不存在表明,聚合通過鏈終止控制的機理進行。
圖10示出丙交酯單體轉化率相對於時間的半對數標繪圖。省略誘導期。[LA]0=0.104M,[LA]0/[初始]0=50,T=90℃,苯-d6。使用[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]、L-丙交酯(紅色線)和外消旋-丙交酯(黑色短劃線)的聚合。
使用[η8-(Pn*)Ti(O-2,6-Me-C6H3)2]的L-丙交酯的聚合在80℃至100℃的溫度範圍內進行並且結果與艾林標繪圖核對。從艾林標繪圖獲得活化參數的估計:ΔH#=75.15kJ mol-1,ΔS#=-125.85J K-1mol-1,ΔG#(100℃)=87.74kJ mol-1。ΔH#的適度的值對於對與金屬中心配位的羰基的攻擊是典型的並且ΔS#的負的和相對高的值意味著在過渡態中的高的有序度。同樣地,所有參數與展現出高度有序的過渡態的配位插入機理一致。L-丙交酯的聚合產生等規立構PLA並且外消旋-丙交酯產生無規立構PLA。
(III)
在圖11中示出了使用[(EBI)Zr(O-2,6-Me-C6H3)Cl]、[(Ind)2Zr(OtBu)Me]和[(Ind)2Zr(O-2,6-Me-C6H3)Me]的L-丙交酯的聚合的偽一級動力學數據。所有最初的聚合研究在80℃下在氯仿-d1中、以50:1的L-LA:引發劑的比率進行,確保[LA]0–0.50M。
圖12示出L-丙交酯轉化率相對於時間的半對數標繪圖,[LA]0/[初始]0=50,[LA]0=0.50M,T=80℃,氯仿-d1(0.5mL),使用[(EBI)Zr(O-2,6-Me-C6H3)Cl](點劃線)、[(Ind)2Zr(OtBu)Me](實線)、[(Ind)2Zr(O-2,6-C6H3)Me](短劃線)的聚合。
圖12示出了絡合物[(Ind)2Zr(OtBu)Me]在八個小時內展現出以84%轉化率的最高活性(k觀察=0.24h-1)並且沒有引發周期。這與在文獻中的鋯引發劑的適中的活性是可比較的。明顯地較低的活性通過[(Ind)2Zr(O-2,6-Me-C6H3)Me]展現,該[(Ind)2Zr(O-2,6-Me-C6H3)Me]在28小時之後、以4.5倍慢於[(Ind)2Zr(OtBu)Me]的速率實現74%轉化率(k觀察=0.05h-1)。感興趣地,[(Ind)2Zr(O-2,6-Me-C6H3)Me]展現出約半小時的引發周期。相比於[(Ind)2Zr(OtBu)Me]的叔丁氧基取代基(tert-butoxide substituent),這些觀察結果通過[(Ind)2Zr(O-2,6-Me-C6H3Me]的芳氧基取代基(aryl-oxidesubstituent)的增加的體積被合理說明。相比之下,[(EBI)Zr(O-2,6-Me-C6H3)Cl]展現出最低的催化活性,在28小時內達成4.2%轉化率(k觀察=0.002h-1),這與由Ning等人,Organometallics,2008,27,5632合成的外消旋-[(EBI)Zr(OC{OiPr}=CMe2)](在18小時內在80℃下在甲苯中的7%轉化率)是可比較的。[(EBI)Zr(O-2,6-Me-C6H3)Cl]的速率比[(Ind)2Zr(OtBu)Me]和[(Ind)2Zr(O-2,6-Me-C6H3)Me]小兩個數量級,可能是由於與叔丁氧基相比,芳氧基的增加的體積以及通過柄型-橋接的配體賦予的增強的剛度二者,防止了茚基部分的再定向。
因為[(Ind)2Zr(OtBu)Me]示出最高的聚合速率,所以進行另外的研究以調查其立體選擇性並且評估其活化。使用[(Ind)2Zr(OtBu)Me]作為引發劑的聚合在60℃和100℃之間以相同的LA:引發劑的比率和如先前使用的[LA]0來進行。活化焓和活化熵從In(k觀察/T)相對於(1/T)的標繪圖(圖12)來計算,給出和這些值與文獻是一致的;對於雙分子反應和配位-插入機理是典型的。
此外,經發現,L-LA的聚合(k觀察=0.24h-1)是在80℃下以50:1的類似的[LA]0:[2]0的比率利用[(Ind)2Zr(OtBu)Me](圖13)的外消旋-LA(k觀察=0.11h-1)的兩倍。這可能是由於在金屬中心處翻轉手性構型所需要的能量屏障,該能量屏障指示鏈終止的控制機制。
使用[(Ind)2Zr(OtBu)Me]作為引發劑的外消旋-LA的聚合的1H{1H}NMR光譜證明了朝向等規立構的PLA的偏向,72%的Pi。
在80℃下,在添加以化學計量的量的叔丁醇與[(Ind)2Zr(OtBu)Me]的情況下,重複L-LA和外消旋-LA的聚合。叔丁醇的添加對L-LA和外消旋-LA兩者的k觀察幾乎不具有影響。對於沒有醇的L-LA的聚合速率與具有醇的L-LA的聚合速率類似(分別地,0.24h-1的k觀察和0.23h-1的k觀察)。對於外消旋-LA的聚合的速率也是類似的(分別地,0.11h-1的k觀察和0.10h-1的k觀察)。具有和不具有叔丁醇的L-LA和外消旋-LA的聚合的分子量和多分散性在表5中被核對。作為引發劑,[(Ind)2Zr(OtBu)Me]證明了L-LA和外消旋-LA在80℃下以50:1的LA:引發劑的比率在氯仿-d1中的高度受控制的聚合,如通過低的多分散性(1.08<Mw/Mn<1.12)所示出的。叔丁醇的添加不影響多分散性;然而,在醇的存在下,實驗分子量顯得更受控制,如不死聚合(immortal polymerisation)所預期的。
表5.用於使用2a的L-丙交酯和外消旋-丙交酯的聚合數據
a聚合條件;[LA]0/[2]0=50,[LA]0=0.5M,80℃。
通過GPC利用聚苯乙烯標準物在THF中測量的。Mn,理論=[LA]0/[2]0x MLAx轉化率。