一種光活性六氫喹啉酮及其製備方法與流程
2023-04-26 18:42:36 2
本發明涉及一種光活性六氫喹啉酮及其製備方法。
背景技術:
光活性六氫喹啉酮不僅是許多具有生物活性分子的重要組成模塊,也是許多天然產物的合成前體。
目前在有機合成中製備光活性六氫喹啉酮的方法主要有多取代吡啶的催化氫化,含疊氮側鏈不飽和酮的重排反應,多組分環化反應等。
技術實現要素:
本發明的目的是提供一種光活性六氫喹啉酮及其製備方法,本發明提供的製備方法,底物價格低廉、反應條件溫和、操作簡便、具有工業化生產潛力的一鍋法合成光活性六氫喹啉酮的方法。
本發明所提供的光活性六氫喹啉酮,其為式Ⅰ所示化合物和式II所示化合物組成的非對映異構體;式Ⅰ所示化合物的非對映異構體過量值為83%~92%;
式Ⅰ和式II中,R1選自下述基團中的任意一種:烷基、吡啶基、噻吩基、萘基、苯基和取代苯基;所述取代苯基中為單取代苯基或雙取代苯基,所述取代苯基中的取代基為甲基、甲氧基、三氟甲基、氟原子、氯原子和溴原子中任一種;
R2為烷基;
R3為氫原子或甲基。
本發明提供的光活性六氫喹啉酮,其中的式I和式II所示化合物具體如下述所示:
式Ⅰ所示化合物的非對映異構體過量值具體可為83%、86%、88%或92%。
本發明進一步提供了所述光活性六氫喹啉酮的製備方法,包括如下步驟:
1)在手性鹼的催化條件下,親核試劑與式III所示不飽和酮酯進行邁克加成反應;
所述親核試劑為1,3-環己二酮或5,5-二甲基-1,3-環己二酮;
式III中,R1和R2的定義同式Ⅰ中;
所述手性鹼的結構式如式IV、式V或式VI所示,
式IV、式V和式VI中,R4為取代苯基或苯乙基,所述取代苯基為單取代或雙取代,所述 取代苯基中的取代基為氫原子、甲基、乙基或三氟甲基;R5為乙基或乙烯基;
2)向步驟1)的反應體系中加入式VII所示苄胺進行反應;
式VII中,R6為氫原子、烷基、烷氧基、三氟甲基、氟原子、氯原子、溴原子或硝基;
3)向步驟2)的反應體系中加入有機強鹼進行反應,得到所述光活性六氫喹啉酮。
上述的製備方法中,步驟1)中,所述邁克加成反應在有機溶劑中進行;所述有機溶劑選自下述任意一種:二氯甲烷、三氯甲烷、1,2-二氯乙烷、甲苯、乙腈和乙酸乙酯;
所述邁克加成反應的反應溫度為0~50℃;反應時間為0.2~10小時。
上述的製備方法中,步驟1)中,所述親核試劑與所述不飽和酮酯的摩爾比為1~3:1;所述手性鹼與所述不飽和酮酯的摩爾比為5~0.1:10;
所述邁克加成反應的體系中,所述不飽和酮酯的濃度為0.1~1.0摩爾/升。
上述的製備方法中,步驟2)中,所述式VII所示苄胺與所述不飽和酮酯的摩爾比為1~10:1;
步驟2)中所述反應的溫度為20~80℃,時間為4~24小時。
上述的製備方法中,步驟3)中,所述有機強鹼為1,8-二氮雜二環十一碳-7-烯或1,5-二氮雜雙環壬-5-烯。
上述的製備方法中,步驟3)中,所述有機強鹼與所述不飽和酮酯的摩爾比為1~2:1~10。
上述的製備方法中,步驟3)中所述反應的溫度為20~80℃,時間為4~24小時。
上述的製備方法中,步驟3)還包括從所述反應液中分離得到式I和式II所示化合物的步驟,具體操作如下:將所述反應液濃縮後進行柱層析純化,經過柱層析後可分別得到光活性六氫喹啉酮的兩個非對映異構體式I和式II。
所述柱層析純化中,洗脫劑可採用二氯甲烷和丙酮體積比為80:1~10:1的混合溶劑;
所用柱的規格具體可為直徑1cm×高15cm;
所用柱填料為200-300目的矽膠。
本發明採用麥可甲醇反應、轉氨化、環化串聯反應策略,以普通易得的手性鹼為催化劑,以不同結構的1,3-環己二酮、不飽和酮酯為底物,苄胺為氮源,有機強鹼促進轉氨化環化反應,採用一鍋法高產率、高對映選擇性及非對映選擇性合成光活性六氫喹啉酮。此方法的原料價格低廉,反應條件溫和,底物使用範圍廣,具有較大的工業化潛力。
本發明提供的光活性六氫喹啉酮,具有手性環狀氨基酯結構,在本發明化合物不飽和酮結構基礎上進行衍生反應可以合成多種具有不同取代基的環狀氨基酯類化合物,因此這些結構多樣的手性環狀氨基酯具有潛在的生物活性,可以為一些藥物的合成提供有用的前體結構。 另外,當手性環狀氨基酯發生水解反應後,可以生成相應的手性環狀胺基酸,而胺基酸在生物體維持正常運轉不可或缺的重要成分,所以相應的具有新穎結構的環狀胺基酸可能會參與和影響生物體信息的複製、轉移和表達等過程,從而表現出不同尋常的生物活性。
附圖說明
圖1為式1所示化合物的合成路線圖。
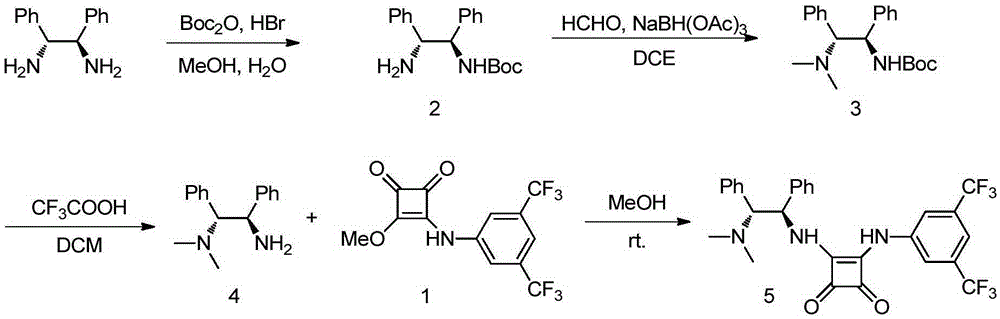
圖2為式5所示催化劑的合成路線圖。
圖3為式III-a不飽和酮酯原料的合成路線圖。
圖4為光活性六氫喹啉酮式I-a和式II-a的合成路線圖。
圖5為III-b不飽和酮酯原料的合成路線圖。
圖6為光活性六氫喹啉酮式I-b和式II-b的合成路線圖。
圖7為III-c不飽和酮酯原料的合成路線圖。
圖8為光活性六氫喹啉酮式I-c和式II-c的合成路線圖。
圖9為III-d不飽和酮酯原料的合成路線圖。
圖10為光活性六氫喹啉酮式I-d和式II-d的合成路線圖。
圖11為III-e不飽和酮酯原料的合成路線圖。
圖12為光活性六氫喹啉酮式I-e和式II-e的合成路線圖。
圖13為III-f不飽和酮酯原料的合成路線圖。
圖14為光活性六氫喹啉酮式I-f和式II-f的合成路線圖。
圖15為III-g不飽和酮酯原料的合成路線圖。
圖16為光活性六氫喹啉酮式I-g和式II-g的合成路線圖。
圖17為III-h不飽和酮酯原料的合成路線圖。
圖18為光活性六氫喹啉酮式I-h和式II-h的合成路線圖。
圖19為III-i不飽和酮酯原料的合成路線圖。
圖20為光活性六氫喹啉酮式I-i和式II-i的合成路線圖。
圖21為光活性六氫喹啉酮式I-j和式II-j的合成路線圖。
圖22為III-j不飽和酮酯原料的合成路線圖。
圖23為光活性六氫喹啉酮式I-k和式II-k的合成路線圖。
具體實施方式
下述實施例中所使用的實驗方法如無特殊說明,均為常規方法。
下述實施例中所用的材料、試劑等,如無特殊說明,均可從商業途徑得到。
下述實施例中所使用的式1所示化合物按照按照圖1所示的合成路線製備,包括下述步驟:
向反應器中加入3,5-二三氟甲基苯胺(11mmol,2.52g),3,4-二甲氧基-3-環丁烯-1,2-二酮(10mmol,1.42g),MeOH(14mL),室溫攪拌48h。所得白色固體過濾,並用少量MeOH 淋洗,乾燥後的白色固體式1(1.80g)所示產物,收率53%。
結構確證結果如下:1H NMR(400MHz,DMSO)δ11.17(s,1H),8.03(s,2H),7.75(s,1H),4.41(s,3H).;13C NMR(100MHz,DMSO)δ187.4,184.5,179.9,169.1,140.2,131.7,131.3,131.0,130.7,124.4,121.7,119.3,116.2,60.9.
下述實施例中所使用的式5所示化合物按照按照圖2所示的合成路線製備,包括下述步驟:
向反應器中加入(1R,2R)-1,2-二苯基乙二胺(17.0mmol,3.60g),45%HBr溶液(1.9mL),H2O(5.0mL),Boc2O(18.7mmol,4.08g),室溫攪拌過夜。加入H2O(40mL),將生成的白色固體濾除,所得濾液濃縮後加入1N NaOH水溶液調節pH>14,於冰水浴中攪拌2h,過濾得到白色固體,將其分散於正己烷(80mL)中,共沸蒸餾除水,得白色固體式2(2.24g)所示產物,收率42%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.40-7.19(m,10H),5.79(s,1H),4.85(s,1H),4.33(s,1H),1.50-1.03(m,11H);13C NMR(100MHz,CDCl3)δ155.9,142.5,141.2,128.7,128.5,127.6,127.4,127.0,126.7,79.5,60.2,28.5.
向反應器中加入式2所示所示白色固體(7.2mmol,2.24g),37%福馬林溶液(35.9mmol,2.9mL),1,2-二氯乙烷(90mL),室溫攪拌15min後加入NaBH(OAc)3(16.6mmol,3.51g),室溫攪拌過夜。加入飽和NaHCO3溶液攪拌30min後,二氯甲烷(50mL×3)萃取水相,合併有機相,Na2SO4乾燥。過濾濃縮後快速柱層析(洗脫劑:DCM:MeOH=50:1),得白色固體式3所示產物1.37g,收率56%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.25-7.13(m,3H),7.12-6.95(m,7H),6.26(s,1H),4.84(s,1H),3.54(d,J=10.7Hz,1H),2.15(s,6H),1.36(s,9H);13C NMR(100MHz,CDCl3)δ156.1,142.1,133.0,129.9,127.9,127.8,127.6,127.5,126.7,79.4,74.1,55.8,40.9,28.5.
向反應器中加入式3所示白色固體1.37g,加入DCM(46mL),CF3COOH(10.2mL),室溫攪拌過夜。濃縮後,加入1N NaOH水溶液調節pH>10,二氯甲烷(20mL×3)萃取水相,合併有機相,Brine洗滌有機相,無水MgSO4乾燥。過濾濃縮後快速柱層析(洗脫劑:DCM:MeOH=30:1),得無色油狀液體式4所示產物0.85g,收率88%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.35-6.97(m,10H),4.43(d,J=10.4Hz,1H),3.64(d,J=10.4Hz,1H),2.22(s,6H),2.03(s,2H).;13C NMR(100MHz,CDCl3)δ143.5,134.1,130.0,128.3,128.2,127.5,127.1,127.0,75.5,55.8,41.2.
向反應器中加入式1所示白色固體(3.54mmol,1.20g),式4所示無色油狀液體(3.54mmol,0.85g),MeOH(18mL),室溫攪拌過夜。將所得白色固體用少量冷的MeOH淋洗,乾燥後得到白色固體式5所示產物1.48g,收率76%。
結構確證結果如下:1H NMR(400MHz,DMSO)δ10.29(s,1H),8.44(s,1H),8.06(s,2H),7.66(s,1H),7.31-7.06(m,10H),5.78(t,J=9.7Hz,1H),4.19(d,J=11.3Hz,1H),2.13(s,6H).; 13C NMR(100MHz,DMSO)δ184.5,180.2,169.0,162.6,141.0,140.5,132.4,131.8,131.4,131.1, 130.8,129.5,128.3,127.7,127.6,127.3,127.2,124.5,121.8,118.2,114.8,71.0,57.9,40.5.
下述實施例中所使用的化合物12按照包括下述步驟的方法製備得到:
將溴代丙酮酸(11.7g)溶於二氯甲烷(100mL)中,轉移至300mL封管中,加入叔丁醇(8.8mL),冷卻至-40℃,緩慢加入濃H2SO4(1.9mL),異丁烯(40mL),封好後移至rt攪拌4d。冷卻至-40℃,打開封管,將溶液倒入100mL飽和NaHCO3水溶液中,待氣體逸完後,二氯甲烷(100mL×3)萃取水相,合併有機相,Brine洗滌有機相至中性,無水MgSO4乾燥。過濾濃縮後所得黃色油狀液體11.08g,與三苯基膦(52.2mmol,13.68g),四氫呋喃(450mL)加入到反應器中,60℃油浴中攪拌2h。冷卻後減壓除去多餘溶劑,加入二氯甲烷(60mL),水(90mL),氫氧化鉀水溶液(2M,26mL),室溫下劇烈攪拌30min後分液,水相用二氯甲烷(50mL×3)萃取,合併有機相,飽和氯化鈉溶液洗滌有機相,無水MgSO4乾燥。過濾,濃縮後加入乙醚100mL,劇烈攪拌,所得固體過濾,真空乾燥後,得到11.73g淺黃色固體式12所示,產率58%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.71-7.61(m,6H),7.60-7.53(m,3H),7.50-7.43(m,6H),1.53(s,9H);13C NMR(100MHz,CDCl3)δ175.6,165.4,165.2,133.4,133.3,132.64,132.61,129.2,129.1,126.3,125.4,81.3,56.2,55.1,28.2.
實施例1、的製備。
1)按照圖3所示的合成路線製備III-a所示不飽和酮酯原料,具體操作如下:
在氮氣氛圍下向反應器中加入苯甲醛(200mmol,21.2g),MeOH(10mL),丙酮酸(200mmol,17.6g),移至0℃低溫反應浴,邊攪拌邊滴加KOH(200mmol,11.2g)的MeOH(45mL)溶液,大約30min加完。加畢,一次性加入KOH(100mmol,5.6g),移至40℃油浴攪拌1h,出現大量黃色固體。後移至0℃反應浴攪拌過夜。次日,將所得黃色固體用布氏漏鬥過濾,冷的MeOH(200mL)淋洗,抽乾後,繼續用油泵真空乾燥,得到黃色固體式7-a所示產物29.5g,收率69%。
於40℃油浴中邊攪拌邊加入水至黃色固體式7-a(29.5g)所示產物全部溶解,將此溶液倒入2N HCl(aq.)(150mL)中,生成大量固體,用DCM/Et2O混合溶劑(200mL×3)萃取水相,合併有機相,無水MgSO4乾燥。過濾濃縮後用乙醚重結晶,過濾後得黃色晶體式8-a(12.5g)所示產物,收率52%。
將黃色晶體式8-a(12.5g)溶於DCM(20mL)中,轉移至300mL封管中,加入叔丁醇(6.0mL),冷卻至-40℃,緩慢加入濃H2SO4(1.4mL),異丁烯(40mL),封好後移至rt攪拌12h。冷卻至-40℃,打開封管,將溶液倒入100mL飽和NaHCO3水溶液中,待氣體逸完後,Et2O(100mL×3)萃取水相,合併有機相,Brine洗滌有機相至中性,無水MgSO4乾燥。過濾濃縮後快速柱層析(洗脫劑:PE:EA=50:1),得黃色油狀液體式III-a所示產物8.45g,收率51%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.80(d,J=16.2Hz,1H),7.66-7.59(m,2H),7.46-7.40(m,3H),7.29(d,J=16.3Hz,1H),1.61(s,9H);13C NMR(100MHz,CDCl3)δ184.3,162.0,148.1,134.3,131.6,129.3,129.1,121.0,84.3,28.1.
2)按照圖4所示的合成路線製備式I-a和式II-a所示化合物,具體操作如下:
向反應器中依次加入1,3-環己二酮(式9-a所示)(0.5mmol,0.056g),底物III-a(0.5mmol,0.116g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為二氯甲烷:丙酮=15/1,v/v),分別得到式I-a和II-a所示光活性六氫喹啉酮,其中式I-a所示的光活性六氫喹啉酮的非對映體過量92%,對映體過量94%。
HPLC條件:手性AD-H柱,流動相:正己烷和異丙醇的體積比為80:20的混合溶劑,流速:1.0mL/min,吸收波長:286nm。
式I-a結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.31-7.24(m,2H),7.21-7.10(m,3H),5.18(br s,1H),4.27(d,J=4.0Hz,1H),3.59(dd,J=12.4,3.6Hz,1H),2.58-2.44(m,2H),2.44-2.29(m,2H),2.23(d,J=12.8Hz,1H),2.08-1.99(m,2H),1.83(td,J=12.8,5.2Hz,1H),1.43(s,9H);13C NMR(100MHz,CDCl3)δ194.0,171.7,158.1,145.1,128.5,128.0,126.3,106.8,83.0,50.2,36.9,34.5,32.5,29.6,28.1,22.0.
經結構鑑定所製備的化合物確為式I-a所示化合物。
式II-a結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.24-7.17(m,2H),7.17-7.07(m,3H),5.06(br s,1H),4.03(t,J=6.4Hz,1H),3.89-3.82(m,1H),2.57-2.16(m,6H),2.06-1.96(m,2H),1.20(s,9H);13C NMR(100MHz,CDCl3)δ194.0,170.9,158.5,145.0,128.4,127.5,126.0,107.9,82.4,52.8,37.1,35.9,34.3,29.8,27.9,21.4。
經結構鑑定所製備的化合物確為式II-a所示化合物。
實施例2、的製備。
1)按照圖5所示的合成路線製備III-b所示不飽和酮酯原料,具體操作如下:
在氮氣氛圍下向反應器中加入2-萘甲醛(100mmol,15.6g),MeOH(20mL),丙酮酸(100mmol,8.8g),移至0℃低溫反應浴,邊攪拌邊滴加KOH(100mmol,5.6g)的MeOH(25mL)溶液,大約30min加完。加畢,一次性加入KOH(50mmol,2.8g),移至40℃油浴攪拌1h,出現大量黃色固體。後移至0℃反應浴攪拌過夜。次日,將所得黃色固體用布氏漏鬥過濾,冷的MeOH(100mL)淋洗,抽乾後,繼續用油泵真空乾燥,得到黃色固體式7-b所示產物24.7 g,收率94%。
於40℃油浴中邊攪拌邊加入水至黃色固體式7-b(24.7g)所示產物全部溶解,將此溶液倒入2N HCl(aq.)(100mL)中,生成大量固體,用DCM/Et2O混合溶劑(200mL×3)萃取水相,合併有機相,無水MgSO4乾燥。過濾濃縮後用石油醚/乙酸乙酯混合溶劑重結晶,過濾後得黃色晶體式8-b(7.24g)所示產物,收率34%。
將黃色晶體式8-b(7.24g)溶於DCM(100mL)中,轉移至300mL封管中,加入叔丁醇(2.8mL),冷卻至-40℃,緩慢加入濃H2SO4(0.6mL),異丁烯(20mL),封好後移至rt攪拌48h。冷卻至-40℃,打開封管,將溶液倒入100mL飽和NaHCO3水溶液中,待氣體逸完後,DCM(100mL×3)萃取水相,合併有機相,Brine洗滌有機相至中性,無水MgSO4乾燥。過濾濃縮後快速柱層析(洗脫劑:PE:EA=40:1),得黃色固體式III-b所示產物4.76g,收率53%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ8.01(s,1H),7.95(d,J=16.1Hz,1H),7.90-7.81(m,3H),7.74(d,J=8.6Hz,1H),7.59-7.50(m,2H),7.40(d,J=16.0Hz,1H),1.63(s,9H).;13C NMR(100MHz,CDCl3)δ184.2,162.0,148.1,134.9,133.4,131.90,131.86,129.1,129.0,128.1,128.0,127.1,123.7,121.0,84.2,28.1.
2)按照圖6所示的合成路線製備式I-b和式II-b所示化合物,具體操作如下:
向反應器中依次加入1,3-環己二酮(式9-a所示)(0.5mmol,0.056g),底物III-b(0.5mmol,0.141g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為二氯甲烷:丙酮=15/1,v/v),分別得到式I-b和II-b所示光活性六氫喹啉酮,其中式I-b所示的光活性六氫喹啉酮的非對映體過量92%,對映體過量92%。
HPLC條件:手性AD-H柱,流動相:正己烷和異丙醇的體積比為70:30的混合溶劑,流速:1.0mL/min,吸收波長:286nm。
式I-b結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.82-7.73(m,3H),7.47-7.37(m,4H),5.19(br s,1H),4.43(d,J=4.0Hz,1H),3.62(dd,J=12.4,3.6Hz,1H),2.66-2.50(m,2H),2.49-2.33(m,2H),2.30(d,J=12.8Hz,1H),2.16-2.03(m,2H),1.89(td,J=12.6,5.2Hz,1H),1.40(s,9H);13C NMR(100MHz,CDCl3)δ194.0,171.7,158.2,142.7,133.6,132.5,128.4,128.0,127.7,127.0,126.2,126.0,125.4,106.7,83.0,50.1,37.0,34.8,32.4,29.6,28.1,22.1.
經結構鑑定所製備的化合物確為式I-b所示化合物。
式II-b結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.76-7.68(m,3H),7.51(s,1H),7.41-7.30(m,3H),5.25(br s,1H),4.19(t,J=6.4Hz,1H),3.88-3.81(m,1H),2.59-2.22(m,6H),2.11-1.94(m,2H),1.02(s,9H);13C NMR(100MHz,CDCl3)δ194.0,170.8,158.8,142.4,133.8,132.5,127.89,127.87,127.6,126.4,125.9,125.7,125.0,107.6,82.3,52.7,37.0,35.9,33.9,29.7,27.6,21.5.
經結構鑑定所製備的化合物確為式II-b所示化合物。
實施例3、的製備。
1)按照圖7所示的合成路線製備III-c所示不飽和酮酯原料,具體操作如下:
在氮氣氛圍下向反應器中加入2,4-二氯苯甲醛(100mmol,17.5g),MeOH(10mL),丙酮酸(100mmol,8.8g),移至0℃低溫反應浴,邊攪拌邊滴加KOH(100mmol,5.6g)的MeOH(25mL)溶液,大約30min加完。加畢,一次性加入KOH(50mmol,2.8g),移至40℃油浴攪拌1h,出現大量黃色固體。後移至0℃反應浴攪拌過夜。次日,將所得黃色固體用布氏漏鬥過濾,冷的MeOH(100mL)淋洗,抽乾後,繼續用油泵真空乾燥,得到黃色固體式7-c所示產物27.8g,收率98%。
於40℃油浴中邊攪拌邊加入水至黃色固體式7-c(27.8g)所示產物全部溶解,將此溶液倒入2N HCl(aq.)(100mL)中,生成大量固體,用DCM/Et2O混合溶劑(200mL×3)萃取水相,合併有機相,無水MgSO4乾燥。過濾濃縮後用石油醚/乙醚混合溶劑重結晶,過濾後得黃色晶體式8-c(7.52g)所示產物,收率31%。
將黃色晶體式8-c(7.52g)溶於DCM(100mL)中,轉移至300mL封管中,加入叔丁醇(2.5mL),冷卻至-40℃,緩慢加入濃H2SO4(0.5mL),異丁烯(20mL),封好後移至rt攪拌48h。冷卻至-40℃,打開封管,將溶液倒入100mL飽和NaHCO3水溶液中,待氣體逸完後,DCM(100mL×3)萃取水相,合併有機相,Brine洗滌有機相至中性,無水MgSO4乾燥。過濾濃縮後快速柱層析(洗脫劑:PE:EA=40:1),得黃色固體式III-c所示產物2.86g,收率31%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ8.11(d,J=16.2Hz,1H),7.65(d,J=8.5Hz,1H),7.47-7.44(m,1H),7.29(d,J=8.4Hz,1H),7.19(d,J=16.2Hz,1H),1.60(s,9H).;13CNMR(100MHz,CDCl3)δ184.2,161.7,142.4,137.7,136.7,131.1,130.4,128.7,127.9,123.7,84.6,28.1.
2)按照圖8所示的合成路線製備式I-c、式II-c所示化合物,具體操作如下:
向反應器中依次加入1,3-環己二酮(式9-a所示)(0.5mmol,0.056g),底物III-c(0.5mmol,0.151g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為二氯甲烷:丙酮=15/1,v/v),分別得到式I-c、II-c所示光活性六氫喹啉酮,其中式I-c所示的光活性六氫喹啉酮的非對映體過量88%,對映體 過量91%。
HPLC條件:手性AD-H柱,流動相:正己烷和異丙醇的體積比為70:30的混合溶劑,流速:1.0mL/min,吸收波長:286nm。
式I-c結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.40(d,J=2.0Hz,1H),7.09(dd,J=8.4,2.0Hz,1H),6.84(d,J=8.4Hz,1H),5.19(br s,1H),4.54(d,J=4.0Hz,1H),3.50(dd,J=12.4,3.6Hz,1H),2.57-2.46(m,2H),2.34(t,J=6.4Hz,2H),2.27(d,J=13.2Hz,1H),2.07-1.98(m,2H),1.80(td,J=12.8,5.6Hz,1H),1.44(s,9H);13C NMR(100MHz,CDCl3)δ193.6,171.3,158.6,140.7,134.5,132.7,130.1,130.0,126.8,106.1,83.4,50.2,36.9,31.9,29.9,29.5,28.1,21.9.
經結構鑑定所製備的化合物確為式I-c所示化合物。
式II-c結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.33(s,1H),7.03(d,J=8.4Hz,1H),6.90(d,J=8.4Hz,1H),5.06(br s,1H),4.38-4.29(m,1H),3.92-3.83(m,1H),2.59-2.44(m,2H),2.39-2.23(m,4H),2.11-1.95(m,2H),1.22(s,9H);13C NMR(100MHz,CDCl3)δ193.6,170.8,158.9,140.4,134.7,132.3,129.6,129.3,127.1,107.8,82.7,52.7,37.0,32.6,30.5,29.8,27.9,21.5.
經結構鑑定所製備的化合物確為式II-c所示化合物。
實施例4、的製備。
1)按照圖9所示的合成路線製備III-d所示不飽和酮酯原料,具體操作如下:
在氮氣氛圍下向反應器中加入4-氟苯甲醛(100mmol,12.4g),MeOH(10mL),丙酮酸(100mmol,8.8g),移至0℃低溫反應浴,邊攪拌邊滴加KOH(100mmol,5.6g)的MeOH(25mL)溶液,大約30min加完。加畢,一次性加入KOH(50mmol,2.8g),移至40℃油浴攪拌1h,出現大量黃色固體。後移至0℃反應浴攪拌過夜。次日,將所得黃色固體用布氏漏鬥過濾,冷的MeOH(100mL)淋洗,抽乾後,繼續用油泵真空乾燥,得到黃色固體式7-d所示產物15.9g,收率68%。
於40℃油浴中邊攪拌邊加入水至黃色固體式7-d(15.9g)所示產物全部溶解,將此溶液倒入2N HCl(aq.)(100mL)中,生成大量固體,用DCM/Et2O混合溶劑(200mL×3)萃取水相,合併有機相,無水MgSO4乾燥。過濾濃縮後用石油醚/乙醚混合溶劑重結晶,過濾後得黃色晶體式8-d(5.77g)所示產物,收率43%。
將黃色晶體式8-d(5.77g)溶於DCM(60mL)中,轉移至300mL封管中,加入叔丁醇(2.6mL),冷卻至-40℃,緩慢加入濃H2SO4(0.6mL),異丁烯(20mL),封好後移至rt攪拌48h。冷卻至-40℃,打開封管,將溶液倒入100mL飽和NaHCO3水溶液中,待氣體逸完後,DCM(100 mL×3)萃取水相,合併有機相,Brine洗滌有機相至中性,無水MgSO4乾燥。過濾濃縮後快速柱層析(洗脫劑:PE:EA=40:1),得黃色固體式III-d所示產物5.05g,收率68%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.76(d,J=16.1Hz,1H),7.65-7.59(m,2H),7.22(d,J=16.1Hz,1H),7.16-7.08(m,2H),1.60(s,9H);13C NMR(100MHz,CDCl3)δ184.0,166.1,163.5,161.9,146.7,131.2,131.1,130.7,120.7,116.7,116.4,84.4,28.1.
2)按照圖10所示的合成路線製備式I-d、式II-d所示化合物,具體操作如下:
向反應器中依次加入5,5-二甲基-1,3-環己二酮(式9-b所示)(0.5mmol,0.071g),底物III-d(0.5mmol,0.125g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為二氯甲烷:丙酮=25/1,v/v),分別得到式I-d、II-d所示光活性六氫喹啉酮,其中式I-d所示的光活性六氫喹啉酮的非對映體過量86%,對映體過量98%。
HPLC條件:手性OD-H柱,流動相:正己烷和異丙醇的體積比為85:15的混合溶劑,流速:1.0mL/min,吸收波長:286nm。
式I-d結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.14-7.05(m,2H),7.00-6.91(m,2H),5.10(br s,1H),4.23(d,J=4.0Hz,1H),3.59(dd,J=12.4,3.6Hz,1H),2.36(s,2H),2.26(d,J=16.4Hz,1H),2.20(d,J=16.4Hz,1H),2.21-2.13(m,1H),1.81(td,J=12.4,4.8Hz,1H),1.44(s,9H),1.14(s,3H),1.09(s,3H);13C NMR(100MHz,CDCl3)δ193.5,171.5,162.9,160.5,156.3,141.1,141.0,129.4,129.3,115.4,115.2,105.5,83.1,50.6,50.1,43.2,33.9,32.8,28.9,28.6,28.1;
經結構鑑定所製備的化合物確為式I-d所示化合物。
式II-d結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.13-7.04(m,2H),6.95-6.84(m,2H),4.95(br s,1H),4.00(t,J=5.6Hz,1H),3.91-3.83(m,1H),2.43-2.15(m,6H),1.22(s,9H),1.16(s,3H),1.08(s,3H);13C NMR(100MHz,CDCl3)δ193.6,170.8,162.7,160.3,156.8,140.69,140.66,129.13,129.05,115.2,115.0,106.4,82.5,52.8,50.7,43.4,35.1,34.0,32.3,29.3,28.4,27.9;
經結構鑑定所製備的化合物確為式II-d所示化合物。
實施例5、的製備。
1)按照圖11所示的合成路線製備III-e所示不飽和酮酯原料,具體操作如下:
在氮氣氛圍下向反應器中加入4-氯苯甲醛(100mmol,14.1g),MeOH(22mL),丙酮酸(100mmol,8.8g),移至0℃低溫反應浴,邊攪拌邊滴加KOH(100mmol,5.6g)的MeOH(25mL)溶液,大約30min加完。加畢,一次性加入KOH(50mmol,2.8g),移至40℃油浴攪拌1h,出現大量黃色固體。後移至0℃反應浴攪拌過夜。次日,將所得黃色固體用布氏漏鬥過濾,冷的MeOH(100mL)淋洗,抽乾後,繼續用油泵真空乾燥,得到黃色固體式7-e所示產物17.1g,收率69%。
於40℃油浴中邊攪拌邊加入水至黃色固體式7-e(17.1g)所示產物全部溶解,將此溶液倒入2N HCl(aq.)(100mL)中,生成大量固體,用DCM/Et2O混合溶劑(200mL×3)萃取水相,合併有機相,無水MgSO4乾燥。過濾濃縮後用石油醚/乙酸乙酯混合溶劑重結晶,過濾後得黃色晶體式8-e(6.99g)所示產物,收率48%。
將黃色晶體式8-e(6.99g)溶於DCM(60mL)中,轉移至300mL封管中,加入叔丁醇(2.9mL),冷卻至-40℃,緩慢加入濃H2SO4(0.6mL),異丁烯(20mL),封好後移至rt攪拌48h。冷卻至-40℃,打開封管,將溶液倒入100mL飽和NaHCO3水溶液中,待氣體逸完後,DCM(100mL×3)萃取水相,合併有機相,Brine洗滌有機相至中性,無水MgSO4乾燥。過濾濃縮後快速柱層析(洗脫劑:PE:EA=40:1),得黃色固體式III-e所示產物5.08g,收率57%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.74(d,J=16.1Hz,1H),7.55(d,J=8.5Hz,2H),7.40(d,J=8.5Hz,2H),7.27d,J=16.1Hz,1H),1.60(s,9H).13C NMR(100MHz,CDCl3)δ183.9,161.8,146.4,137.7,132.9,130.2,129.6,121.4,84.4,28.1.
2)按照圖12所示的合成路線製備式I-e、式II-e所示化合物,具體操作如下:
向反應器中依次加入5,5-二甲基-1,3-環己二酮(式9-b所示)(0.5mmol,0.071g),底物III-e(0.5mmol,0.133g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為二氯甲烷:丙酮=25/1,v/v),分別得到式I-e、II-e所示光活性六氫喹啉酮,其中式I-e所示的光活性六氫喹啉酮的非對映體過量86%,對映體過量98%。
HPLC條件:手性OD-H柱,流動相:正己烷和異丙醇的體積比為85:15的混合溶劑,流速:1.0mL/min,吸收波長:286nm。
式I-e結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.24(d,J=8.4Hz,2H),7.08(d,J=8.4Hz,2H),5.13(br s,1H),4.21(d,J=3.6Hz,1H),3.58(dd,J=12.4,3.6Hz,1H),2.36(s,2H),2.26(d,J=16.4Hz,1H),2.20(d,J=16.4Hz,1H),2.21-2.14(m,1H),1.82(td,J=12.4,5.2Hz,1H),1.44(s,9H),1.13(s,3H),1.09(s,3H);13C NMR(100MHz,CDCl3)δ193.5,171.4,156.4,144.0,132.1,129.4,128.7,105.2,83.2,50.5,50.1,43.1,34.1,32.8,32.6,28.9,28.6,28.1;
經結構鑑定所製備的化合物確為式I-e所示化合物。
式II-e結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.17(d,J=8.4Hz,2H),7.06(d,J=8.4Hz,2H),5.03(br s,1H),3.98(t,J=6.4Hz,1H),3.90-3.82(m,1H),2.43-2.25(m,3H), 2.23-2.13(m,3H),1.22(s,9H),1.15(s,3H),1.08(s,3H);13C NMR(100MHz,CDCl3)δ193.6,170.7,157.0,143.7,131.6,129.1,128.5,106.0,82.6,52.8,50.6,43.3,35.4,33.9,32.3,29.2,28.3,27.8;
經結構鑑定所製備的化合物確為式II-e所示化合物。
實施例6、的製備。
1)按照圖13所示的合成路線製備III-f所示不飽和酮酯原料,具體操作如下:
在氮氣氛圍下向反應器中加入4-溴苯甲醛(100mmol,18.5g),MeOH(10mL),丙酮酸(100mmol,8.8g),移至0℃低溫反應浴,邊攪拌邊滴加KOH(100mmol,5.6g)的MeOH(25mL)溶液,大約30min加完。加畢,一次性加入KOH(50mmol,2.8g),移至40℃油浴攪拌1h,出現大量黃色固體。後移至0℃反應浴攪拌過夜。次日,將所得黃色固體用布氏漏鬥過濾,冷的MeOH(100mL)淋洗,抽乾後,繼續用油泵真空乾燥,得到黃色固體式7-f所示產物24.0g,收率82%。
於40℃油浴中邊攪拌邊加入水至黃色固體式7-f(24.0g)所示產物全部溶解,將此溶液倒入2N HCl(aq.)(100mL)中,生成大量固體,用DCM/Et2O混合溶劑(200mL×3)萃取水相,合併有機相,無水MgSO4乾燥。過濾濃縮後用乙醚重結晶,過濾後得黃色晶體式8-f(8.93g)所示產物,收率43%。
將黃色晶體式8-f(8.93g)溶於DCM(100mL)中,轉移至300mL封管中,加入叔丁醇(3.1mL),冷卻至-40℃,緩慢加入濃H2SO4(0.7mL),異丁烯(20mL),封好後移至rt攪拌48h。冷卻至-40℃,打開封管,將溶液倒入100mL飽和NaHCO3水溶液中,待氣體逸完後,DCM(100mL×3)萃取水相,合併有機相,Brine洗滌有機相至中性,無水MgSO4乾燥。過濾濃縮後快速柱層析(洗脫劑:PE:EA=40:1),得黃色固體式III-f所示產物2.49g,收率23%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.72(d,J=16.0Hz,1H),7.56(d,J=8.8Hz,2H),7.48(d,J=8.4Hz,2H),7.28(d,J=16.0Hz,1H),1.60(s,9H).13C NMR(100MHz,CDCl3)δ183.9,161.8,146.5,133.3,132.6,130.4,126.1,121.4,84.4,28.1.
2)按照圖14所示的合成路線製備式I-f、式II-f所示化合物,具體操作如下:
向反應器中依次加入5,5-二甲基-1,3-環己二酮(式9-b所示)(0.5mmol,0.071g),底物III-f(0.5mmol,0.156g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌 10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為二氯甲烷:丙酮=25/1,v/v),分別得到式I-f、II-f所示光活性六氫喹啉酮,其中式I-f所示的光活性六氫喹啉酮的非對映體過量83%,對映體過量97%。
HPLC條件:手性OD-H柱,流動相:正己烷和異丙醇的體積比為85:15的混合溶劑,流速:1.0mL/min,吸收波長:286nm。
式I-f結構確證結果如下:1H NMR(400MHz,CDCl3)δδ7.39(d,J=8.4Hz,2H),7.02(d,J=8.0Hz,2H),5.13(br s,1H),4.19(d,J=4.0Hz,1H),3.58(dd,J=12.4,3.6Hz,1H),2.35(s,2H),2.26(d,J=16.4Hz,1H),2.20(d,J=16.4Hz,1H),2.20-2.13(m,1H),1.81(td,J=12.4,5.2Hz,1H),1.44(s,9H),1.13(s,3H),1.09(s,3H);13C NMR(100MHz,CDCl3)δ193.5,171.4,156.4,144.5,131.6,129.8,120.2,105.1,83.2,50.5,50.1,43.1,34.2,32.8,32.5,28.9,28.6,28.1;
經結構鑑定所製備的化合物確為式I-f所示化合物。
式II-f結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.32(d,J=8.0Hz,2H),7.01(d,J=8.0Hz,2H),5.06(br s,1H),3.96(t,J=6.4Hz,1H),3.89-3.83(m,1H),2.41-2.25(m,3H),2.22-2.13(m,3H),1.22(s,9H),1.15(s,3H),1.08(s,3H);13C NMR(100MHz,CDCl3)δ193.5,170.7,157.1,144.2,131.4,129.5,119.7,105.9,82.6,52.7,50.6,43.3,35.4,33.8,32.3,29.2,28.3,27.8;
經結構鑑定所製備的化合物確為式II-f所示化合物。
實施例7、的製備。
1)按照圖15所示的合成路線製備III-g所示不飽和酮酯原料,具體操作如下:
向封管中加入4-三氟甲基苯甲醛(5mmol,0.87g),化合物12(7.5mmol,3.03g),1,2-二氯乙烷(20mL),移至60℃油浴,攪拌2d。濃縮後快速柱層析(洗脫劑:PE:EA=20:1),得黃色固體式III-g所示產物0.76g,收率51%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.79(d,J=16.2Hz,1H),7.71(d,J=8.1Hz,2H),7.67(d,J=8.1Hz,2H),7.36(d,J=16.2Hz,1H),1.60(s,9H)..13C NMR(100MHz,CDCl3)δ183.8,161.5,145.7,137.7,133.0,132.7,130.4,129.1,126.2,126.17,125.2,123.1,122.5,84.6,28.1.
2)按照圖16所示的合成路線製備式I-g、式II-g所示化合物,具體操作如下:
向反應器中依次加入5,5-二甲基-1,3-環己二酮(式9-b所示)(0.5mmol,0.071g),底物III-g(0.5mmol,0.150g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為石油醚:乙酸乙酯=1/1,v/v),分別得到式I-g、II-g所示光活性六氫喹啉酮,其中式I-g所示的光活性六氫喹啉酮的非對映體過量83%,對映體過量95%。
HPLC條件:手性OD-H柱,流動相:正己烷和異丙醇的體積比為92:8的混合溶劑,流速:1.5mL/min,吸收波長:286nm。
式I-g結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.53(d,J=7.6Hz,2H),7.26(d,J=8.0Hz,2H),5.20(br s,1H),4.29(d,J=4.4Hz,1H),3.58(dd,J=12.4,3.6Hz,1H),2.38(s,2H),2.30-2.18(m,3H),1.87(td,J=12.4,5.2Hz,1H),1.44(s,9H),1.14(s,3H),1.10(s,3H);13C NMR(100MHz,CDCl3)δ193.5,171.2,156.7,149.6,129.1,128.8,128.6,128.5,128.3,128.2,125.9,125.61,125.57,125.53,125.50,123.2,120.5,104.9,83.3,50.5,50.1,43.1,34.7,32.7,32.4,28.9,28.6,28.1;
經結構鑑定所製備的化合物確為式I-g所示化合物。
式II-g結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.47(d,J=8.0Hz,2H),7.25(d,J=8.8Hz,2H),4.99(br s,1H),4.07(t,J=6.4Hz,1H),3.93-3.87(m,1H),2.46-2.28(m,3H),2.24-2.16(m,3H),1.20(s,9H),1.18(s,3H),1.10(s,3H);13C NMR(100MHz,CDCl3)δ193.5,170.5,157.6,149.4,128.7,128.6,128.3,128.1,128.0,127.7,125.9,125.32,125.28,125.24,125.21,123.2,120.5,105.2,82.5,52.6,50.6,43.2,35.6,33.3,32.3,29.1,28.3,27.7;
經結構鑑定所製備的化合物確為式II-g所示化合物。
實施例8、的製備。
1)按照圖17所示的合成路線製備III-h所示不飽和酮酯原料,具體操作如下:
在氮氣氛圍下向反應器中加入4-甲氧基苯甲醛(100mmol,18.5g),MeOH(10mL),丙酮酸(100mmol,8.8g),移至0℃低溫反應浴,邊攪拌邊滴加KOH(100mmol,5.6g)的MeOH(25mL)溶液,大約30min加完。加畢,一次性加入KOH(50mmol,2.8g),移至40℃油浴攪拌1h,出現大量黃色固體。後移至0℃反應浴攪拌過夜。次日,將所得黃色固體用布氏漏鬥過濾,冷的MeOH(100mL)淋洗,抽乾後,繼續用油泵真空乾燥,得到黃色固體式7-g所示產物20.0g,收率82%。
於40℃油浴中邊攪拌邊加入水至黃色固體式7-g(20.0g)所示產物全部溶解,將此溶 液倒入2NHCl(aq.)(100mL)中,生成大量固體,用DCM/Et2O混合溶劑(200mL×3)萃取水相,合併有機相,無水MgSO4乾燥。過濾濃縮後用乙醚/石油醚重結晶,過濾後得黃色晶體式8-g(8.92g)所示產物,收率53%。
將黃色晶體式8-g(8.92g)溶於DCM(100mL)中,轉移至300mL封管中,加入叔丁醇(3.8mL),冷卻至-40℃,緩慢加入濃H2SO4(0.8mL),異丁烯(20mL),封好後移至rt攪拌48h。冷卻至-40℃,打開封管,將溶液倒入100mL飽和NaHCO3水溶液中,待氣體逸完後,DCM(100mL×3)萃取水相,合併有機相,Brine洗滌有機相至中性,無水MgSO4乾燥。過濾濃縮後快速柱層析(洗脫劑:PE:EA=20:1),得黃色固體式III-g所示產物2.80g,收率25%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.76(d,J=16.1Hz,1H),7.58(d,J=8.8Hz,2H),7.16(d,J=16.1Hz,1H),6.93(d,J=8.8Hz,2H),3.86(s,3H),1.60(s,9H).13C NMR(100MHz,CDCl3)δ184.2,162.6,162.3,148.0,131.1,127.1,118.7,114.8,84.1,55.7,28.1.
2)按照圖18所示的合成路線製備式I-h、式II-h所示化合物,具體操作如下:
向反應器中依次加入5,5-二甲基-1,3-環己二酮(式9-b所示)(0.5mmol,0.071g),底物III-h(0.5mmol,0.131g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為二氯甲烷:丙酮=25/1,v/v),分別得到式I-h、II-h所示光活性六氫喹啉酮,其中式I-h所示的光活性六氫喹啉酮的非對映體過量88%,對映體過量94%。
HPLC條件:手性OD-H柱,流動相:正己烷和異丙醇的體積比為70:30的混合溶劑,流速:1.0mL/min,吸收波長:286nm。
式I-h結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.06(d,J=8.4Hz,2H),6.82(d,J=8.4Hz,2H),5.09(br s,1H),4.20(d,J=3.6Hz,1H),3.76(s,3H),3.63(dd,J=12.0,3.2Hz,1H),2.35(s,2H),2.29-2.16(m,3H),1.78(td,J=12.8,5.2Hz,1H),1.43(s,9H),1.13(s,3H),1.09(s,3H);13C NMR(100MHz,CDCl3)δ193.5,171.8,158.2,156.1,137.5,128.9,114.0,105.9,83.0,55.4,50.6,50.3,43.2,33.8,32.8,32.7,28.9,28.7,28.2;
經結構鑑定所製備的化合物確為式I-h所示化合物。
式II-h結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.04(d,J=8.8Hz,2H),6.75(d,J=8.8Hz,2H),4.94(br s,1H),3.97(t,J=6.4Hz,1H),3.88-3.84(m,1H),3.73(s,3H),2.42-2.26(m,3H),2.26-2.16(m,3H),1.21(s,9H),1.16(s,3H),1.08(s,3H);13C NMR(100MHz,CDCl3)δ193.6,171.0,157.9,156.6,137.1,128.6,113.9,106.8,82.3,55.4,52.9,50.7,43.4,35.0,34.2,32.3,29.4,28.3,27.9;
經結構鑑定所製備的化合物確為式II-h所示化合物。
實施例9、的製備。
1)按照圖19所示的合成路線製備III-i所示不飽和酮酯原料,具體操作如下:
在氮氣氛圍下向反應器中加入4-甲基苯甲醛(100mmol,12.0g),MeOH(10mL),丙酮酸(100mmol,8.8g),移至0℃低溫反應浴,邊攪拌邊滴加KOH(100mmol,5.6g)的MeOH(25mL)溶液,大約30min加完。加畢,一次性加入KOH(50mmol,2.8g),移至40℃油浴攪拌1h,出現大量黃色固體。後移至0℃反應浴攪拌過夜。次日,將所得黃色固體用布氏漏鬥過濾,冷的MeOH(100mL)淋洗,抽乾後,繼續用油泵真空乾燥,得到黃色固體式7-i所示產物19.8g,收率87%。
於40℃油浴中邊攪拌邊加入水至黃色固體式7-i(9.10g)所示產物全部溶解,將此溶液倒入2N HCl(aq.)(44mL)中,生成大量固體,用DCM/Et2O混合溶劑(50mL×3)萃取水相,合併有機相,無水MgSO4乾燥。過濾濃縮後用乙醚/石油醚重結晶,過濾後得黃色晶體式8-i(4.98g)所示產物,收率66%。
將黃色晶體式8-i(4.98g)溶於乙醚(100mL)中,轉移至300mL封管中,加入叔丁醇(2.2mL),冷卻至-40℃,緩慢加入濃H2SO4(0.5mL),異丁烯(15mL),封好後移至rt攪拌48h。冷卻至-40℃,打開封管,將溶液倒入100mL飽和NaHCO3水溶液中,待氣體逸完後,DCM(100mL×3)萃取水相,合併有機相,Brine洗滌有機相至中性,無水MgSO4乾燥。過濾濃縮後快速柱層析(洗脫劑:PE:EA=40:1),得黃色固體式III-i所示產物1.63g,收率25%。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.77(d,J=16.1Hz,1H),7.52(d,J=7.9Hz,2H),7.28-7.20(m,3H),2.39(s,3H),1.60(s,9H).13C NMR(100MHz,CDCl3)δ184.3,162.1,148.3,142.4,131.6,130.0,129.2,120.0,84.2,28.1,21.8.
2)按照圖20所示的合成路線製備式I-i、式II-i所示化合物,具體操作如下:
向反應器中依次加入5,5-二甲基-1,3-環己二酮(式9-b所示)(0.5mmol,0.071g),底物III-i(0.5mmol,0.123g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為二氯甲烷:丙酮=40/1,v/v),分別得到式I-i、II-i所示光活性六氫喹啉酮,其中式I-i所示的光活性六氫喹啉酮的非對映體過量86%,對映體過量96%。
HPLC條件:手性OD-H柱,流動相:正己烷和異丙醇的體積比為70:30的混合溶劑,流速:1.0mL/min,吸收波長:286nm。
式I-i結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.11-7.01(m,4H),5.09(br s,1H),4.21(d,J=3.6Hz,1H),3.66(dd,J=12.8,4.0Hz,1H),2.35(s,2H),2.30(s,3H),2.25(d,J=16.4Hz,1H),2.20(d,J=16.4Hz,1H),2.21-2.17(m,1H),1.79(td,J=12.4,4.8Hz,1H),1.43(s,9H),1.13(s,3H),1.09(s,3H);13C NMR(100MHz,CDCl3)δ193.5,171.8,156.1,142.4,135.7,129.2,127.9,105.8,83.0,50.6,50.3,43.2,34.2,32.7,32.6,28.9,28.7,28.2,21.2;
經結構鑑定所製備的化合物確為式I-i所示化合物。
式II-i結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.02(s,4H),5.01(br s,1H),3.96(t,J=6.8Hz,1H),3.89-3.82(m,1H),2.43-2.26(m,3H),2.25(s,3H),2.21-2.13(m,3H),1.22(s,9H), 1.16(s,3H),1.08(s,3H);13C NMR(100MHz,CDCl3)δ193.5,170.9,156.7,142.2,135.3,129.1,127.5,106.7,82.3,52.9,50.7,43.3,35.8,34.6,32.2,29.4,28.2,27.8,21.2;
經結構鑑定所製備的化合物確為式II-i所示化合物。
實施例10、的製備。
1)按照圖7所示的合成路線製備III-c所示不飽和酮酯原料,具體操作如實施例3中所述。
2)按照圖21所示的合成路線製備式I-j、式II-j所示化合物,具體操作如下:
向反應器中依次加入5,5-二甲基-1,3-環己二酮(式9-b所示)(0.5mmol,0.071g),底物III-c(0.5mmol,0.151g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為二氯甲烷:丙酮=25/1,v/v),分別得到式I-j、II-j所示光活性六氫喹啉酮,其中式I-j所示的光活性六氫喹啉酮的非對映體過量86%,對映體過量97%。
HPLC條件:手性AD-H柱,流動相:正己烷和異丙醇的體積比為80:20的混合溶劑,流速:1.0mL/min。吸收波長:286nm。
式I-j結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.40(d,J=2.0Hz,1H),7.10(dd,J=8.0,2.0Hz,1H),6.84(d,J=8.4Hz,1H),5.16(br s,1H),4.55(d,J=4.4Hz,1H),3.53(dd,J=12.4,3.6Hz,1H),2.43-2.31(m,2H),2.29-2.18(m,3H),1.79(td,J=12.8,5.6Hz,1H),1.44(s,9H),1.12(s,3H),1.08(s,3H);13C NMR(100MHz,CDCl3)δ193.2,171.3,156.9141.0,134.5,132.7,130.04,130.01,126.7,104.8,83.4,50.5,50.3,43.1,32.7,31.8,30.1,29.1,28.4,28.1;
經結構鑑定所製備的化合物確為式I-j所示化合物。
式II-j結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.33(s,1H),7.02(d,J=8.0Hz,1H),6.87(d,J=8.0Hz,1H),5.16(br s,1H),4.41-4.26(m,1H),3.95-3.81(m,1H),2.51-2.24(m,4H),2.18(s,2H),1.20(s,9H),1.15(s,3H),1.08(s,3H);13C NMR(100MHz,CDCl3)δ193.1,170.7,157.5,140.5,134.7,132.2,129.7,129.3,127.0,105.4,82.6,52.7,50.5,43.3,32.4,30.2,29.0,28.6,27.8;
經結構鑑定所製備的化合物確為式II-j所示化合物。
實施例11、的製備。
1)按照圖22所示的合成路線製備III-j所示不飽和酮酯原料,具體操作如下:
向封管中加入正丁醛(40mmol,2.88g),化合物12(10mmol,4.04g),1,2-二氯乙烷(40mL),移至70℃油浴,攪拌1d。濃縮後快速柱層析(洗脫劑:PE:Et2O=40:1),得黃色固體式III-j所示產物0.44g,22%yield。
結構確證結果如下:1H NMR(400MHz,CDCl3)δ7.14-7.03(m,1H),6.53(d,J=16.0Hz,1H),2.27(q,J=7.1Hz,2H),1.65-1.49(m,11H),0.94(t,J=7.3Hz,3H).13C NMR(100MHz,CDCl3)δ184.9,162.4,154.5,125.7,84.1,35.2,28.121.3,13.9.
2)按照圖23所示的合成路線製備式I-k、式II-k所示化合物,具體操作如下:
向反應器中依次加入5,5-二甲基-1,3-環己二酮(式9-b所示)(0.5mmol,0.071g),底物III-j(0.5mmol,0.099g),手性鹼催化劑(式5所示)(0.0125mmol,0.0068g)和1.0mL ClCH2CH2Cl,室溫攪拌3h。向反應器中加入4Cl-PhCH2NH2(1.5mmol,0.2124g)將反應器放入50℃的油浴中,攪拌12小時後,加入DBU(0.25mmol,0.038g),繼續於50℃攪拌10h。將反應器從油浴取出,冷卻後旋幹,柱層析(洗脫劑為二氯甲烷:丙酮=25/1,v/v),分別得到式I-k、II-k所示光活性六氫喹啉酮,其中式I-k所示的光活性六氫喹啉酮的非對映體過量83%,對映體過量51%。
HPLC條件:手性OD-H柱,流動相:正己烷和異丙醇的體積比為95:5的混合溶劑,流速:1.0mL/min,吸收波長:286nm。
式I-k結構確證結果如下:1H NMR(400MHz,CDCl3)δ5.01(br s,1H),3.88(dd,J=12.4,3.6Hz,1H),2.97-2.88(m,1H),2.30-2.11(m,5H),1.58-1.39(m,3H),1.50(s,9H),1.38-1.25(m,1H),1.20-1.08(m,1H),1.04(s,3H),1.03(s,3H),0.93(t,J=7.2Hz,3H);13C NMR(100MHz,CDCl3)δ193.8,172.1,155.0,108.5,82.7,50.7,50.4,43.1,36.8,32.5,28.5,28.2,28.13,28.08,20.3,14.4;
經結構鑑定所製備的化合物確為式I-k所示化合物。
式II-k結構確證結果如下:1H NMR(400MHz,CDCl3)δ4.84(br s,1H),3.84(s,1H),2.84-2.75(m,1H),2.31-2.12(m,5H),2.03-1.91(m,1H),1.65-1.19(m,3H),1.47(s,9H),1.15-1.01(m,7H),0.88(t,J=7.2Hz,3H);13C NMR(100MHz,CDCl3)δ194.2,172.1,155.4,108.7,82.4,52.6,51.0,43.5,35.7,32.3,29.1,28.8,28.1,27.9,27.6,20.6,14.3;
經結構鑑定所製備的化合物確為式II-k所示化合物。