一種溫控型變黏酸及其製備方法與流程
2023-05-06 07:16:56 3
本發明屬於化學合成技術領域,尤其涉及一種溫控型變黏酸及其製備方法。
背景技術:
酸化作業中要利用酸液的化學溶蝕作用及向地層擠酸時的水力作用,解除油層堵塞,擴大和連通油氣層孔隙,恢復和提高油氣層滲透率。大量的作業資料證實,確保注入的酸液是否完全覆蓋整個待處理層是酸化作用成功與否的關鍵。但是非均質碳酸鹽巖地層各個裂縫的滲透率存在差異,當酸液注入地層後,會優先進入滲透率大的地層裂縫,而滲透率較低的層段得不到處理。這些未得到處理的層段意味著產量較少,並損失一定的儲量。為此,人們開發出了使酸液轉向的機械和化學方法,以確保碳酸鹽巖儲層能夠得到一致的增產處理,變黏酸作為一種化學轉向方法得到了較為廣泛的應用。根據使用的稠化劑不同變黏酸可以分為兩大類:聚合物變黏酸和黏彈性表面活性劑變黏酸;按照變黏機理不同又可以分為:溫度控制變黏酸和pH值控制變黏酸。目前,在油田現場有過應用或文獻有過報導的變黏酸主要有三種:聚合物溫控變黏酸、聚合物pH值控制變黏酸、黏彈性表面活性劑pH值控制變黏酸。
聚合物pH值控制變黏酸又稱降濾失酸(LCA),在保持了稠化酸低摩阻、緩速等優點的基礎上,強化了酸液的濾失控制。LCA具有較低的初始黏度(30~50mpa.s),當pH值升至2~4時,高價金屬離子交聯劑與稠化劑發生交聯反應,使酸液交聯增黏至1000mpa.s以上;當pH值進一步升高,破膠劑將金屬交聯劑還原或螯合形成穩定的化合物,破壞聚合物與金屬陽離子交聯劑形成的交聯體系,使體系破膠黏度降低。LAC酸液體系的使用缺陷是pH值2~4時酸液體系粘度才大幅度升高,而此時酸液已經消耗殆盡,並且不能在高溫環境下使用,高溫條件下酸巖反應過快,可能導致酸液還沒有交聯就消耗完,同時要另外引入金屬離子作為交聯劑會對地層造成二次傷害。
聚合物溫控變黏酸(TCA)是通過酸液體系的溫度變化來控制酸液體系的粘度。常溫條件下酸液粘度較低(低於50mpa.s),泵入地層後,隨著體系溫度升高,酸液中的稠化劑和交聯劑在高溫環境下發生交聯反應,使酸液體系粘度升高。其同樣存在需要引入金屬離子交聯劑造成破膠不徹底,容易對地層造成二次傷害。
黏彈性表面活性劑pH值控制變黏酸是在酸液中添加黏彈性表面活性劑作為稠化劑,依靠表面活性劑之間的締合作用,形成蠕蟲狀或棒狀膠束,膠束之間互相纏結在溶液中形成網狀結構來提高酸液的黏度。現階段應用的黏彈性表面活性劑質量分數均在5%以上,成本較高,同時耐溫性較差。
相對於聚合物類變黏酸易對儲層造成二次傷害,表面活性劑類變黏酸易破膠,低傷害是重要的優勢,也是變黏酸發展的方向。
技術實現要素:
本發明所要解決的技術問題是提供一種溫控型變黏酸及其製備方法,該溫控型變黏酸利用超分子聚合物與表面活性劑之間的相互協同效應實現溫控變黏,避免了常用聚合物變黏酸引入金屬離子交聯劑對地層造成二次傷害等問題。
本發明解決上述技術問題所採用的技術方案是:一種溫控型變黏酸,利用超分子聚合物與表面活性劑的相互協同作用實現溫控變粘,由下列質量百分比的組分組成:鹽酸12~20%,溫控型變黏酸稠化劑0.6~1.5%,溫控型變黏酸活化劑0.3~1.0%,緩蝕劑1~2%,鐵離子穩定劑1~2%,破乳劑1~2%,助排劑1~2%,餘量為水,其中緩蝕劑、鐵離子穩定劑、破乳劑、助排劑選擇配伍性良好的;
所述溫控型變黏酸稠化劑為一種超分子聚合物,由下列質量百分比的組分組成:丙烯醯胺15~20%,陰離子不飽和單體15~20%,兩性離子不飽和單體1~3%,過硫酸銨-亞硫酸氫鈉0.1~0.5%,片鹼2~4%,純鹼1~2%,分散劑3%,其餘為水,其中過硫酸銨與亞硫酸氫鈉的質量比為3:1,所述分散劑為高分子表面活性劑類分散劑;
所述溫控型變黏酸活化劑為一種表面活性劑,由下列質量百分比的組分組成:十六烷基二甲基叔胺16~18%,正十二醇22~27%,離子化合物15~20%,氫氧化鈉1.5~3%,偶氮二異丁腈1~3%,其餘為溶劑甲醇。
進一步的是,由下列質量百分比的組分組成:鹽酸15%,溫控型變黏酸稠化劑0.8%,溫控型變黏酸活化劑0.5%,緩蝕劑2%,鐵離子穩定劑1%,破乳劑1%,助排劑1%,餘量為水。
進一步的是,所述兩性離子不飽和單體為N-n烷基二烯丙基磺基甜菜鹼,其中n為12、14、16或18;陰離子不飽和單體為2-丙烯醯胺基-2-甲基丙磺酸或甲基丙烯酸鈉鹽。
進一步的是,所述N-n烷基二烯丙基磺基甜菜鹼為十六烷基二烯丙基磺基甜菜鹼。
進一步的是,所述高分子表面活性劑類分散劑為聚乙烯基吡咯烷酮。
進一步的是,所述溫控型變黏酸稠化劑由下列質量百分比的組分組成:丙烯醯胺20%,2-丙烯醯胺基-2-甲基丙磺酸15%,十六烷基二烯丙基磺基甜菜鹼3%,過硫酸銨-亞硫酸氫鈉0.2%,片鹼2.1%,純鹼1.5%,聚乙烯基吡咯烷酮3%,其餘為水,其中過硫酸銨與亞硫酸氫鈉的質量比為3:1。
進一步的是,所述離子化合物為2-酮基葡萄糖酸鈣。
進一步的是,所述溫控型變黏酸活化劑由下列質量百分比的組分組成:十六烷基二甲基叔胺16%,正十二醇25%,2-酮基葡萄糖酸鈣18%,氫氧化鈉3%,偶氮二異丁腈1%,其餘為溶劑甲醇。
一種溫控型變黏酸的製備方法,包括以下步驟:
(1)製備溫控型變黏酸稠化劑
取丙烯醯胺、兩性離子不飽和單體、陰離子不飽和單體和水均勻混合得到混合液,先加入片鹼將體系中和至pH值為7左右,然後加入純鹼調節體系pH值至9~10,加入高分子表面活性劑作為分散劑,加入氧化還原引發劑過硫酸銨-亞硫酸氫鈉,在反應溫度為25~30℃下反應4~6h,90℃下水解4h後得到膠體狀產物,對膠體進行造粒、乾燥、粉碎,即得溫控型變黏酸稠化劑;
(2)製備溫控型變黏酸活化劑
取甲醇作為溶劑,加入十六烷基二甲基叔胺、正十二醇和2-酮基葡萄糖酸鈣做為反應物,加入氫氧化鈉調節pH值為8~9,加入催化劑偶氮二異丁腈,在45~55℃條件下,進行酯化反應4~5h,反應結束後,加入活性炭,然後冷卻、過濾、烘乾得到溫控型變黏酸活化劑的固體產物;將溫控型變黏酸活化劑的固體產物加水溶解,配製成質量分數為50%的溶液,溶解完全後,攪拌混合均勻,即得溫控型變黏酸活化劑;
(3)製備溫控型變黏酸
取步驟(1)中製備的溫控型變黏酸稠化劑、步驟(2)中製備的溫控型變黏酸活化劑、鹽酸、緩蝕劑、鐵離子穩定劑、破乳劑、助排劑和水混合均勻,即得溫控型變黏酸。
進一步的是,所述步驟(1)中反應溫度為25℃,反應時間為5h。
本發明的有益效果:本發明製得的壓裂液具有低溫低粘,高溫高粘的特性,實現了溫控變粘,有利於延長酸液的作用距離,實現深部酸化,同時提高了可泵注性,降低了管道摩阻;另外還具有良好的抗溫抗剪切性、緩蝕緩速性,並能徹底破膠。
附圖說明
圖1為本發明一種溫控型變黏酸稠化劑酸溶時間測定曲線;
圖2為本發明一種溫控型變黏酸稠化劑增稠能力測試曲線;
圖3為本發明一種溫控型變黏酸稠化劑抗鹽能力測試曲線;
圖4為本發明一種溫控型變黏酸變黏前對比圖片;
圖5為本發明一種溫控型變黏酸變黏後對比圖片;
圖6為本發明一種溫控型變黏酸120℃耐溫耐剪切測試曲線;
圖7為本發明一種溫控型變黏酸緩速性能測試曲線;
圖8為本發明一種溫控型變黏酸降阻率測試曲線;
圖9為本發明一種溫控型變黏酸現場施工曲線。
具體實施方式
本發明的一種溫控型變黏酸,利用超分子聚合物與表面活性劑的相互協同作用實現溫控變粘,由下列質量百分比的組分組成:鹽酸12~20%,溫控型變黏酸稠化劑0.6~1.5%,溫控型變黏酸活化劑0.3~1.0%,緩蝕劑1~2%,鐵離子穩定劑1~2%,破乳劑1~2%,助排劑1~2%,餘量為水,其中緩蝕劑、鐵離子穩定劑、破乳劑、助排劑選擇配伍性良好的;
所述溫控型變黏酸稠化劑為一種超分子聚合物,由下列質量百分比的組分組成:丙烯醯胺15~20%,陰離子不飽和單體15~20%,兩性離子不飽和單體1~3%,過硫酸銨-亞硫酸氫鈉0.1~0.5%,片鹼2~4%,純鹼1~2%,分散劑3%,其餘為水,其中過硫酸銨與亞硫酸氫鈉的質量比為3:1,所述分散劑為高分子表面活性劑類分散劑;
所述溫控型變黏酸活化劑為一種表面活性劑,由下列質量百分比的組分組成:十六烷基二甲基叔胺16~18%,正十二醇22~27%,離子化合物15~20%,氫氧化鈉1.5~3%,偶氮二異丁腈1~3%,其餘為溶劑甲醇。
進一步的是,由下列質量百分比的組分組成:鹽酸15%,溫控型變黏酸稠化劑0.8%,溫控型變黏酸活化劑0.5%,緩蝕劑2%,鐵離子穩定劑1%,破乳劑1%,助排劑1%,餘量為水。
進一步的是,所述兩性離子不飽和單體為N-n烷基二烯丙基磺基甜菜鹼,其中n為12、14、16或18;陰離子不飽和單體為2-丙烯醯胺基-2-甲基丙磺酸或甲基丙烯酸鈉鹽。
進一步的是,所述N-n烷基二烯丙基磺基甜菜鹼為十六烷基二烯丙基磺基甜菜鹼。
進一步的是,所述高分子表面活性劑類分散劑為聚乙烯基吡咯烷酮。
進一步的是,為了取得產品性能最有優良的溫控型變黏酸稠化劑,所以溫控型變黏酸稠化劑由下列質量百分比的組分組成:丙烯醯胺20%,2-丙烯醯胺基-2-甲基丙磺酸15%,十六烷基二烯丙基磺基甜菜鹼3%,過硫酸銨-亞硫酸氫鈉0.2%,片鹼2.1%,純鹼1.5%,聚乙烯基吡咯烷酮3%,其餘為水,其中過硫酸銨與亞硫酸氫鈉的質量比為3:1。
進一步的是,所述離子化合物為2-酮基葡萄糖酸鈣。
進一步的是,為了取得產品性能最優良的溫控型變黏酸活化劑,所以溫控型變黏酸活化劑由下列質量百分比的組分組成:十六烷基二甲基叔胺16%,正十二醇25%,2-酮基葡萄糖酸鈣18%,氫氧化鈉3%,偶氮二異丁腈1%,其餘為溶劑甲醇。
一種溫控型變黏酸的製備方法,包括以下步驟:
(1)製備溫控型變黏酸稠化劑
取丙烯醯胺、兩性離子不飽和單體、陰離子不飽和單體和水均勻混合得到混合液,先加入片鹼將體系中和至pH值為7左右,然後加入純鹼調節體系pH值至9~10,加入高分子表面活性劑作為分散劑,加入氧化還原引發劑過硫酸銨-亞硫酸氫鈉,在反應溫度為25~30℃下反應4~6h,90℃下水解4h後得到膠體狀產物,對膠體進行造粒、乾燥、粉碎,即得溫控型變黏酸稠化劑;
(3)製備溫控型變黏酸活化劑
取甲醇作為溶劑,加入十六烷基二甲基叔胺、正十二醇和2-酮基葡萄糖酸鈣做為反應物,加入氫氧化鈉調節pH值為8~9,加入催化劑偶氮二異丁腈,在45~55℃條件下,進行酯化反應4~5h,反應結束後,加入活性炭,然後冷卻、過濾、烘乾得到溫控型變黏酸活化劑的固體產物;將溫控型變黏酸活化劑的固體產物加水溶解,配製成質量分數為50%的溶液,溶解完全後,攪拌混合均勻,即得溫控型變黏酸活化劑;
(3)製備溫控型變黏酸
取步驟(1)中製備的溫控型變黏酸稠化劑、步驟(2)中製備的溫控型變黏酸活化劑、鹽酸、緩蝕劑、鐵離子穩定劑、破乳劑、助排劑和水混合均勻,即得溫控型變黏酸。
其中步驟(3)製備溫控型變黏酸的過程本發明提供實驗室內配製方法和施工現場配製方法:
實驗室內配製方法如下:在實驗室內,按變黏酸組分的重量百分比,向燒杯中加入水和鹽酸,放入磁子,然後置於磁力攪拌器上,在攪拌條件下緩慢向溶液中加入變黏酸稠化劑,攪拌均勻後,依次加入變黏酸活化劑、緩蝕劑、鐵離子穩定劑和其它添加劑,繼續攪拌即可得到溫控型變黏酸。
施工現場配製方法如下:在施工現場,按變黏酸組分的重量百分比,在配液罐中加入水和鹽酸,然後加入緩蝕劑、鐵離子穩定劑和其它添加劑,攪拌均勻後,再在大罐攪拌條件下從射流槍吸入變黏酸稠化劑,稠化劑的吸入須緩慢,即以不結塊和不形成魚眼為準;稠化劑加完後,攪拌20min,液體靜置2小時,可得到溫控型變黏酸。
進一步的是,所述步驟(1)中反應溫度為25℃,反應時間為5h。
本發明提供了一種溫控型變黏酸稠化劑和活化劑的合成方法,以及利用該稠化劑和活化劑製備一種溫控型變黏酸的方法,該稠化劑和溫控型變黏酸壓裂液的優良性能主要概述如下:
(1)變黏酸稠化劑的酸液表觀粘度隨使用濃度的增大而增大,在常溫下,這種稠化劑對酸液具有較強的增粘能力;
(2)變黏酸稠化劑分子中帶有疏水基團,在酸中能夠形成一種具有可逆結構的流體,具有良好的粘彈性;
(3)常溫下活化劑可以屏蔽稠化劑部分分子鏈之間的締合作用,聚合物溶液的表觀粘度較低,受熱以後,活化劑在酸液中發生降解,濃度降低,和稠化劑分子鏈的協同效應發生了變化,增強了聚合物分子鏈間的締合作用,形成了更強的空間立體結構,實現酸液的溫控變黏;
(4)正是由於稠化劑與活化劑之間的相互作用,該溫控變黏酸在常溫下粘度較低(30mPa·s~50mPa·s),提高了酸液的泵注性,降低了管道摩阻,隨著溫度的升高,粘度增大,降低了酸巖反應速度,實現深部酸化;
(5)某區油層滲透率低,注水井啟動壓力高,某井停注5小時後,注水量下降,利用該溫控變黏酸酸化措施後近井地帶孔道疏通,注水啟動壓力降低,起到降壓增注目的,七個月內該井累計增注7169m3,獲得了良好的增注效果。
下面結合實施例對本發明的具體實施方式做進一步的描述,並不因此將本發明限制在所述的實施例範圍之中。若未特別指出,實施例中涉及到的壓裂液性能評價實驗條件和實驗方法均參照中石油天然氣行業標準「SY/T6214-1996酸液稠化劑評價方法」;若未特別指出,實施例中涉及到的百分號「%」均為質量百分比。
實施例1
本發明的一種溫控型變黏酸,由下列質量百分比的組分組成:鹽酸12%,溫控型變黏酸稠化劑0.6%,溫控型變黏酸活化劑0.3%,緩蝕劑1%,鐵離子穩定劑1%,破乳劑1%,助排劑1%,餘量為水,其中緩蝕劑、鐵離子穩定劑、破乳劑、助排劑選擇配伍性良好的;
所述溫控型變黏酸稠化劑由下列質量百分比的組分組成:丙烯醯胺20%,陰離子不飽和單體15%,兩性離子不飽和單體3%,過硫酸銨-亞硫酸氫鈉0.2%,片鹼2.1%,純鹼1.5%,分散劑3%,其餘為水,其中過硫酸銨與亞硫酸氫鈉的質量比為3:1,所述分散劑為高分子表面活性劑類分散劑;
所述溫控型變黏酸活化劑由下列質量百分比的組分組成:十六烷基二甲基叔胺16%,正十二醇25%,離子化合物18%,氫氧化鈉3%,偶氮二異丁腈1%,其餘為溶劑甲醇。
實施例2
本發明的一種溫控型變黏酸,由下列質量百分比的組分組成:鹽酸20%,溫控型變黏酸稠化劑1.5%,溫控型變黏酸活化劑1.0%,緩蝕劑2%,鐵離子穩定劑2%,破乳劑2%,助排劑2%,餘量為水,其中緩蝕劑、鐵離子穩定劑、破乳劑、助排劑選擇配伍性良好的;
所述溫控型變黏酸稠化劑由下列質量百分比的組分組成:丙烯醯胺20%,陰離子不飽和單體20%,兩性離子不飽和單體3%,過硫酸銨-亞硫酸氫鈉0.5%,片鹼4%,純鹼2%,分散劑3%,其餘為水,其中過硫酸銨與亞硫酸氫鈉的質量比為3:1,所述分散劑為高分子表面活性劑類分散劑;
所述溫控型變黏酸活化劑由下列質量百分比的組分組成:十六烷基二甲基叔胺18%,正十二醇27%,離子化合物20%,氫氧化鈉3%,偶氮二異丁腈3%,其餘為溶劑甲醇。
實施例3
本發明的一種溫控型變黏酸,由下列質量百分比的組分組成:由下列質量百分比的組分組成:鹽酸15%,溫控型變黏酸稠化劑0.8%,溫控型變黏酸活化劑0.5%,緩蝕劑2%,鐵離子穩定劑1%,破乳劑1%,助排劑1%,餘量為水,其中緩蝕劑、鐵離子穩定劑、破乳劑、助排劑選擇配伍性良好的;
所述溫控型變黏酸稠化劑由下列質量百分比的組分組成:丙烯醯胺20%,2-丙烯醯胺基-2-甲基丙磺酸15%,十六烷基二烯丙基磺基甜菜鹼3%,過硫酸銨-亞硫酸氫鈉0.2%,片鹼2.1%,純鹼1.5%,聚乙烯基吡咯烷酮3%,其餘為水,其中過硫酸銨與亞硫酸氫鈉的質量比為3:1;
所述溫控型變黏酸活化劑由下列質量百分比的組分組成:十六烷基二甲基叔胺16%,正十二醇25%,2-酮基葡萄糖酸鈣18%,氫氧化鈉3%,偶氮二異丁腈1%,其餘為溶劑甲醇。
該實施例的一種溫控型變黏酸的製備方法如下:
(1)製備溫控型變黏酸稠化劑
將丙烯醯胺、N-十六烷基二烯丙基磺基甜菜鹼、2-丙烯醯胺基-2-甲基丙磺酸和水加入反應釜中混合均勻,加入片鹼和純鹼調節pH值,用精密pH值測得pH為9.6,加入分散劑聚乙烯吡咯烷酮,加入氧化還原引發劑過硫酸銨-亞硫酸氫鈉,在溫度為25℃下反應5h,並在90℃下水解4h後得到膠體狀產物,對膠體進行造粒、乾燥、粉碎,即得溫控型變黏酸稠化劑。
(2)製備溫控型變黏酸活化劑
向反應釜中加入甲醇作為溶劑,加入十六烷基二甲基叔胺、正十二醇和2-酮基葡萄糖酸鈣做為反應物,加入片鹼調節pH值,用精密pH試紙測得pH為8.2,加入催化劑偶氮二異丁腈,在50℃條件下進行酯化反應4h,反應結束後,加入活性炭,然後冷卻、過濾、烘乾得到變黏酸用活化劑固體產物。
將得到的溫控型變黏酸活化劑固體產物加水溶解,配製成質量分數為50%的溶液,溶解完全後,攪拌混合均勻,即得溫控型變黏酸活化劑。
(3)製備溫控型變黏酸
取步驟(1)中製備的溫控型變黏酸稠化劑、步驟(2)中製備的溫控型變黏酸活化劑、鹽酸、緩蝕劑、鐵離子穩定劑、破乳劑、助排劑和水混合均勻,即得溫控型變黏酸。
對本發明實施例3中製備的溫控型變黏酸稠化劑進行各個性能測試
1、對上述實施例3中步驟(3)製備過程中溫控型變黏酸稠化劑的酸溶時間進行測定
按重量百分比,步驟(3)的具體過程為:向燒杯中加入共198.4g的水和鹽酸,鹽酸質量分數為20%,放入磁子,然後置於磁力攪拌器上,用磁力攪拌器進行攪拌,調節攪拌速度至溶液剛好形成漩渦(轉速大約為500r/min),稱取步驟(1)中製備好的溫控型變黏酸稠化劑1.6g緩慢均勻加入,攪拌均勻後每隔5min使用六速旋轉粘度計在170S-1下測定酸液的表觀粘度,具體的變化曲線見圖1。本發明製備的溫控型變黏酸稠化劑在酸中的溶解時間較短,為26min左右,有利於現場配液,降低施工成本。
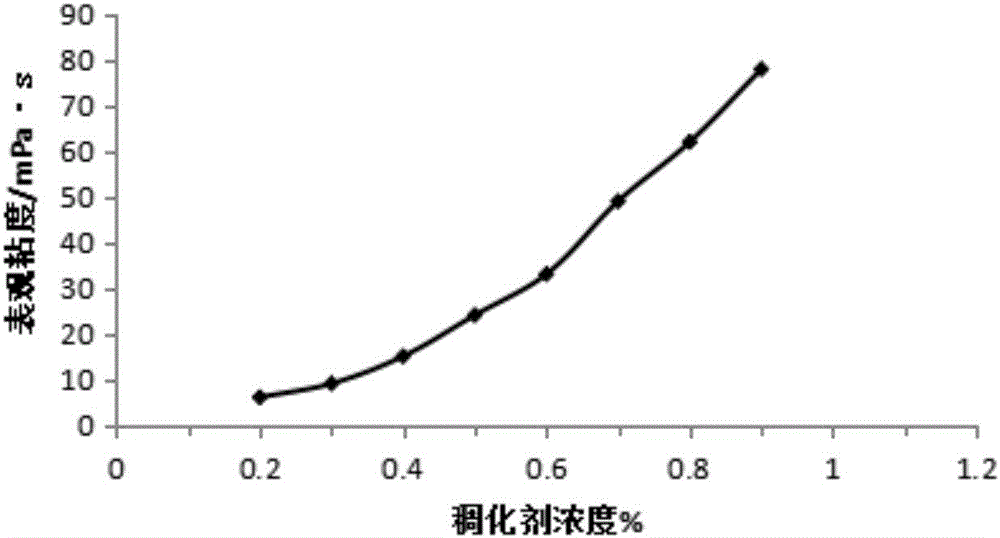
2、對本發明實施例3中步驟(1)製備的溫控型變黏酸稠化劑增稠能力進行測定
本發明提供的溫控變黏酸稠化劑,其20%鹽酸溶液的表觀粘度隨稠化劑使用濃度的增大而增大,具體的變化曲線見圖2。在常溫下,這種溫控變黏酸稠化劑對酸液具有較強的增粘能力。
3、對本發明實施例3中步驟(1)製備的溫控型變黏酸稠化劑抗鹽能力進行測試
按重量百分比,配製溫控型變黏酸稠化劑濃度為0.8%的20%的鹽酸溶液,然後加入一定濃度的CaCl2和NaCl(質量比為1:1),調節加入鹽的總質量濃度,待其溶解均勻後使用六速旋轉粘度計在170S-1下測定鹽酸溶液的表觀粘度,具體的變化曲線見圖3。隨著鹽濃度的增大,酸液粘度緩慢的下降,但是下降幅度不大,表明該稠化劑具有良好的抗鹽性能。
對本發明實施例3中製備的溫控型變黏酸進行各個性能測試
1、本發明實施例3製備得到的溫控型變黏酸前後的對比圖
本發明實施例3提供的一種溫控型變黏酸,經加熱後能夠形成一定強度的凍膠,並具有一定的挑掛性,實驗現象見圖4和圖5,實驗表明該溫控型變黏酸能通過稠化劑與活化劑之間的相互作用來提高強度,從而提高變黏酸的各項性能,實現深部酸化。
2、本發明實施例3製備得到的溫控型變黏酸120℃耐溫耐剪切測試
使用德國Haake公司的RS-600流變儀測定其高溫流變性,鹽酸質量濃度為20%,溫控型變黏酸稠化劑質量濃度為0.8%,溫控型變黏酸活化劑質量濃度為0.5%,然後加入2%的緩蝕劑WLD-31A、1%的鐵離子穩定劑B-180、1%的助排劑DJ-02和1%的破乳劑NE-4240,設定剪切速率為170S-1,測試溫度120℃,時間2h,考察粘度與時間的變化關係如圖6。
3、本發明實施例3製備得到的溫控型變黏酸緩速性能測試
按重量百分比,配製鹽酸濃度為20%、稠化劑濃度為0.8%、活化劑濃度為0.5%、緩蝕劑濃度為2%、鐵離子穩定劑濃度為1%、助排劑濃度為1%和破乳劑濃度為1%的變黏酸,在90℃條件下與碳酸鈣反應,每隔10min取出少量的酸液用標準的氫氧化鈉溶液進行滴定。變黏酸相比於空白酸具有較好的緩速效果,可延緩酸巖反應,有助於實現深部酸化,緩速效果見圖7。
4、本發明實施例3製備得到的溫控型變黏酸腐蝕性能測試
按重量百分比,配製鹽酸濃度為20%、稠化劑濃度為0.8%、活化劑濃度為0.5%、緩蝕劑濃度為2%、鐵離子穩定劑濃度為1%、助排劑濃度為1%和破乳劑濃度為1%的變黏酸,在常壓、90℃條件下測定其腐蝕速度,試驗結果見表1。
表1溫控型變黏酸腐蝕速度測試
測試結果表明:除了緩蝕劑的作用外,變黏酸中聚合物之間的相互作用也降低了H+的擴散速度,這兩種作用確保變黏酸在施工過程中能充分降低對管道的腐蝕,延長管道的使用壽命。
5、本發明實施例3製備得到的溫控型變黏酸破膠性能測試
按重量百分比,配製鹽酸濃度為20%、稠化劑濃度為0.8%、活化劑濃度為0.5%、緩蝕劑濃度為2%、鐵離子穩定劑濃度為1%、助排劑濃度為1%和破乳劑濃度為1%的變黏酸,取200ml在常溫條件下過量的碳酸鈣反應,每隔30min取殘酸測定其表觀粘度,試驗結果見表2。
表2變黏酸的破膠情況
測試結果表明:該變黏酸與碳酸鈣能反應完全,可徹底破膠。最後所得殘酸的密度為1.05g/ml,其表面張力為28.6mN/m。
6、本發明實施例3製備得到的溫控型變黏酸降阻性能測試
按重量百分比,配製鹽酸濃度為20%、稠化劑濃度分別為0.6%~0.8%、活化劑濃度為0.5%、緩蝕劑濃度為2%、鐵離子穩定劑濃度為1%、助排劑濃度為1%和破乳劑濃度為1%的變黏酸。選用8mm的管路進行測試,記錄通過管路後的摩阻壓降來計算減阻率,根據減阻率的大小評價水溶性減阻劑的減阻效果,減阻率與流量的關係如圖8,最大減阻率可達到57%。
減阻率計算:
式中:Φ----減阻率,%;
ΔΡ水----清水通過測試管路時的壓差,MPa;
ΔΡ----含水溶性減阻劑流體通過測試管路時的壓差,MPa。
對本發明實施例3製備得到的溫控型變黏酸進行現場實驗
某95-96井2015年10月25日投注,11月19日連接注水管線停注5小時,開注後該井注水壓力升高,注入量減少,12月27日注不進,2016年1月10日實施酸化處理。複合型變粘酸用量15m3,變黏酸配方為:15%HCL+0.8%稠化劑+0.5%活化劑+2%緩蝕劑+1%鐵離子穩定劑+1%助排劑,最高壓力18MPa,工作壓力9MPa,停泵9MPa,施工曲線見圖9。該區油層滲透率低,注水井啟動壓力高,該井停住5小時後,注水量下降,酸化措施後近井地帶孔道疏通,注水啟動壓力降低,起到降壓增注目的。截止6月25日該井累計增注7169m3。
應當理解的是,對本領域普通技術人員來說,可以根據上述說明加以改進或變換,而所有這些改進和變換都應屬於本發明所附權利要求的保護範圍。