一種分離純化納豆芽孢桿菌中維生素K2的方法與流程
2023-11-06 21:32:01 2
本發明涉及生物工程技術領域,尤其涉及一種分離純化納豆芽孢桿菌中維生素K2的方法。
背景技術:
維生素K2(Menaquinone,MK,VK2)是一類甲萘醌類化合物的統稱,化學結構式為2-甲基-3-烯基-1,4-萘醌,根據C-3側鏈上異戊二烯單元的個數,MK可分為14種,以MK-n表示(n=1-14)。研究表明,維生素K2不僅可以促進血液凝固,增加骨鈣素合成,而且還可降低患肝癌和前列腺癌的風險。最近的研究表明,維生素K2還可用於帕金森氏綜合症及肌肉萎縮性側索硬化症的輔助治療。維生素K2的安全性已經得到中國食品藥品監督管理局、美國藥品研究所、美國FDA及歐盟理事會和歐洲議會的認定。現在,歐盟已將維生素K2作為一種功能性食品,在美國,有關VK2的藥用研究也已進入了臨床三期階段。
隨著我國經濟社會的發展、人民生活水平的提高以及人口進入老齡化階段,高純度的維生素K2無論作為一種保健品還是藥品,都具有廣泛的需求。
維生素K2生產菌株主要包括革蘭氏陽性菌中的納豆芽孢桿菌(Bacilus subtilis)和革蘭氏陰性菌中的黃桿菌屬(Flavobacterium sp.)。其中,納豆芽孢桿菌主要產維生素K2,又因其菌體本身為益生菌,所產的維生素K2也具有較高的產量和較好的生物相容性,因此,是發酵生產維生素K2的優良菌種。
因此,研究適合於從納豆芽孢桿菌發酵液中分離純化維生素K2的一整套工業化方法,對於VK2的產業化,對於改善人民生活質量,具有重要的經濟和社會意義。
根據目前文獻報導,分離純化納豆芽孢桿菌中維生素K2的方法主要或者集中於某一步的研究(Aydin Berenjian et al.Appl Biochem Biotechnol,2014,172:1347-1357;Sato et al.Journal of Industrial Microbiology and Biotechnology,2001,26(3),115-120;Yanagisawa,et al.Journal of Food Biochemistry,2005,29(3),267-277),或者集中於實驗室的樣品檢測(Patrick J.et al.Journal of lipid research,1971,12:442-449;Hiroyuki Wakabayashi et al.Nutrition,2003,19:661-665;)。目前,國內僅有專利CN104262129B公開了一種納豆枯草芽孢桿菌以及由該菌株提純維生素甲萘醌-7的方法。而國外尚未見此類報導。本發明更系統性、詳細地公開了一種從納豆芽孢桿菌發酵液中提取分離純化維生素K2(VK2)的方法。
技術實現要素:
本發明的目的在於克服現有技術的不足,提供一種具備產品純度高、生物活性高、回收率高,分離純化工藝操作簡單、條件溫和、處理能力大,各有機溶劑可回收利用以及便於工業化應用等優點的分離純化納豆芽孢桿菌中維生素K2的方法。
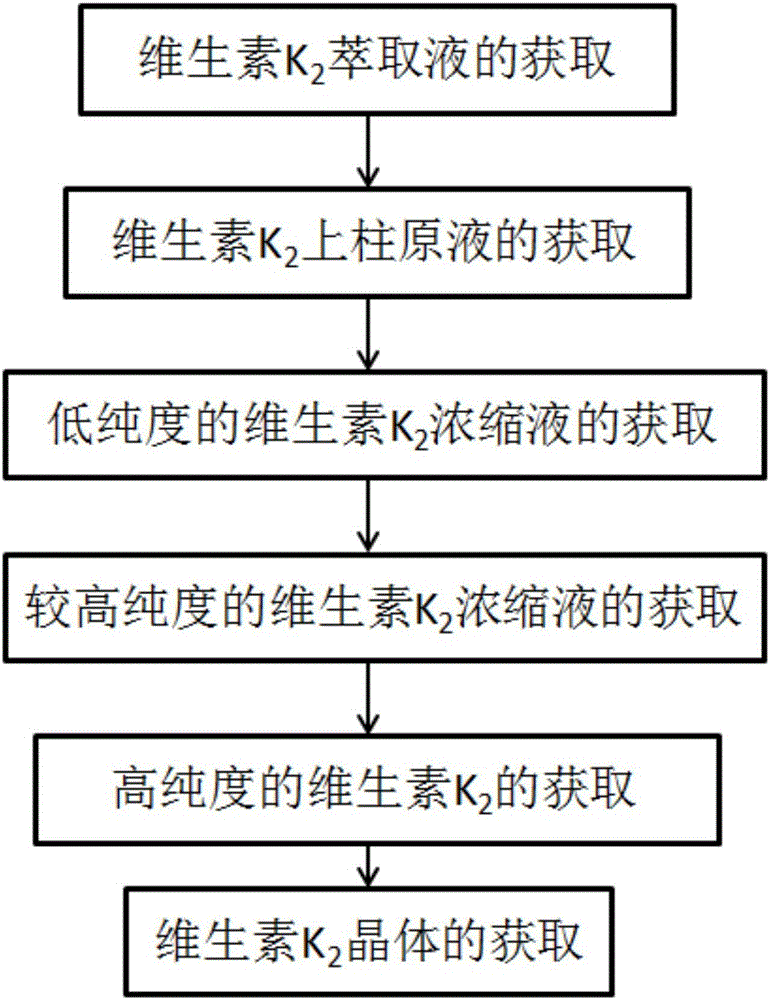
本發明是通過以下技術方案實現的:一種分離純化納豆芽孢桿菌中維生素K2的方法,包括如下步驟:
(1)維生素K2萃取液的獲取
以納豆芽孢桿菌發酵液為維生素K2提取原液,離心獲取菌體,烘乾後,採用第一有機溶劑進行萃取以獲取維生素K2萃取液;
(2)維生素K2上柱原液的獲取
將維生素K2萃取液通過膜分離設備除去不溶性雜質,將獲得的維生素K2濾液蒸乾,然後以極性有機溶劑重溶,獲取維生素K2上柱原液;
(3)低純度的維生素K2濃縮液的獲取
將維生素K2上柱原液通過大孔吸附樹脂柱吸附,採用溼法或幹法上柱,然後以第二有機溶劑清洗,最後用第三有機溶劑進行洗脫,收集相應洗脫液並對其進行濃縮,從而獲取低純度的維生素K2濃縮液;
(4)較高純度的維生素K2濃縮液的獲取
將低純度的維生素K2濃縮液經過分子量排阻層析柱純化,然後以第四有機溶劑進行洗脫,收集相應洗脫液並對其進行濃縮,從而獲取較高純度的維生素K2濃縮液;
(5)高純度的維生素K2的獲取
將較高純度的維生素K2濃縮液溶解於第五有機溶劑後,通過反相矽膠柱分離,然後用第六有機溶劑洗脫並分段收集,即可獲得高純度的維生素K2;
(6)維生素K2晶體的獲取
以第七有機溶劑將維生素K2高溫溶解,低溫結晶,並重結晶,即可獲得維生素K2晶體。
優選地,所述步驟(1)中納豆芽孢桿菌發酵液的組成為:大豆粉65g/L,玉米粉60g/L,蛋白腖50g/L;所述發酵液的發酵周期為7-9天。
優選地,所述步驟(1)中萃取條件為:萃取方式為靜置萃取或者攪拌萃取,萃取時間20min-60min,萃取次數為1-3次,用於萃取的第一有機溶劑為乙醇、正丁醇、乙酸、二氯甲烷、異戊醇、正己烷、乙酸乙酯、三氯甲烷、四氯化碳、丙酮、乙醚、甲苯中的一種或多種的混合液。
優選地,所述步驟(2)中膜分離設備的膜孔徑≤0.45μm,所述極性有機溶劑為甲醇、乙醇、乙二醇、乙腈、丙酮、水中的一種或幾種的混合液。
優選地,所述步驟(3)中大孔吸附樹脂為非極性和/或弱極性大孔吸附樹脂,所述上柱條件為:高徑比為3-10:1,流速為0.025-0.110倍柱體積/min。
優選地,所述大孔吸附樹脂為芳香族吸附劑。
優選地,所述步驟(4)中分子量排阻層析填料所排阻的物質的分子量為400Da-1000Da;所述填料可分別做柱或分幾段裝於同一個分子量排阻層析柱中,層析液按照填料排阻極限由小到大或由大到小的順序依次經過柱填料;所述分子量排阻層析柱高徑比為10-25:1,流速為0.004-0.010倍柱體積/min,層析柱高度與維生素K2濃縮液上液高度比為35-50:1。
優選地,所述步驟(3)中第二有機溶劑為極性有機溶劑,用量為大孔吸附樹脂柱柱體積的1-3倍;所述第三有機溶劑為二氯甲烷、正丁醇、異丁醇、甲苯、庚烷、環戊烷、環己烷、正己烷、石油醚、正戊烷、異戊烷中的一種或幾種的混合液,用量為柱體積的1-4倍;所述步驟(4)中第四有機溶劑為苯、甲苯、四氯化碳、二甲基甲醯胺、酮類、二氯甲烷、二氯代苯、四氯乙烯、四氫呋喃和三氯代苯中的一種或幾種的混合液,其用量為分子量排阻層析柱柱體積的1-4倍。
優選地,所述步驟(5)中第五有機溶劑與第六有機溶劑為極性或弱極性有機溶劑,為甲醇、乙醇、乙二醇、乙腈、丙酮、異丙醇、乙酸乙酯、四氫呋喃、丙醇、乙酸丁酯、正丁醇、二氯甲烷、異丁醇中的一種或幾種的混合液;所述反相矽膠柱的矽膠粒徑為10-80μm,高徑比為5-25:1,流速為0.015-0.065倍柱體積/min,柱高與較高純度的維生素K2濃縮液上液高度比為7-30:1。
優選地,所述步驟(6)中第七有機溶劑為含有一定超純水的甲醇或者乙醇;所述超純水的含量為0-30%;所述的高溫溫度範圍為40-85℃,低溫溫度範圍為-40-0℃。
優選地,所述濃縮方法為減壓濃縮,減壓濃縮溫度為40-99℃,真空度小於-0.08MPa;所述蒸乾方法為減壓蒸乾。
優選地,所述低純度的維生素K2濃縮液的純度為小於50%,較高純度的維生素K2濃縮液的純度為50-80%,高純度的維生素K2的純度為大於80%。
優選地,所述維生素K2晶體的純度為大於或等於98.2%。
優選地,所述維生素K2回收率大於85%。
本發明的優點在於:(1)獲得的維生素K2晶體產品純度高、生物活性高、回收率高;(2)分離純化工藝操作簡單、條件溫和、處理能力大;(3)各有機溶劑可回收利用,便於工業化應用。
附圖說明
圖1是本發明實施例1-3提供的一種分離純化納豆芽孢桿菌中維生素K2的方法的流程圖;
圖2是本發明實施例3提供的一種分離純化納豆芽孢桿菌中維生素K2的方法的不同有機溶劑萃取維生素K2效率圖;
圖3是本發明實施例3提供的一種分離純化納豆芽孢桿菌中維生素K2的方法的發酵樣品萃取液HPLC液相圖譜圖;
圖4是本發明實施例3提供的一種分離純化納豆芽孢桿菌中維生素K2的方法的發酵樣品純化後HPLC液相圖譜圖。
具體實施方式
下面對本發明的實施例作詳細說明,本實施例在以本發明技術方案為前提下進行實施,給出了詳細的實施方式和具體的操作過程,但本發明的保護範圍不限於下述的實施例。
實施例1
一種分離純化納豆芽孢桿菌中維生素K2的方法,如圖1所示,包括如下步驟:
(1)維生素K2萃取液的獲取
以納豆芽孢桿菌發酵液為維生素K2提取原液,離心獲取菌體,烘乾後,採用乙醇進行萃取以獲取維生素K2萃取液;其中萃取方式為靜置萃取,萃取時間20min,萃取次數為1次。
(2)維生素K2上柱原液的獲取
將維生素K2萃取液通過膜分離設備(膜孔徑≤0.45μm)除去不溶性雜質,將獲得的維生素K2濾液減壓蒸乾,然後以甲醇重溶,獲取維生素K2上柱原液;
(3)低純度的維生素K2濃縮液的獲取
將維生素K2上柱原液通過大孔吸附樹脂柱吸附(芳香族吸附劑),採用溼法上柱(上柱條件為:高徑比為3:1,流速為0.025倍柱體積/min),然後以極性有機溶劑清洗(用量為大孔吸附樹脂柱柱體積的1倍),最後用二氯甲烷進行洗脫(用量為大孔吸附樹脂柱柱體積的1倍),收集相應洗脫液並對其進行濃縮,從而獲取低純度的維生素K2濃縮液(純度小於50%);
(4)較高純度的維生素K2濃縮液的獲取
將低純度的維生素K2濃縮液經過分子量排阻層析柱純化,然後以苯進行洗脫(其用量為分子量排阻層析柱柱體積的1倍),收集相應洗脫液並對其進行濃縮,從而獲取較高純度的維生素K2濃縮液(純度為50-80%);
其中,所述分子量排阻層析填料所排阻的物質的分子量為400Da-1000Da;所述填料可分別做柱或分幾段裝於同一個分子量排阻層析柱中,層析液按照填料排阻極限由小到大或由大到小的順序依次經過柱填料;所述分子量排阻層析柱高徑比為10:1,流速為0.004倍柱體積/min,層析柱高度與維生素K2濃縮液上液高度比為35:1;
(5)高純度的維生素K2的獲取
將較高純度的維生素K2濃縮液溶解於甲醇後,通過反相矽膠柱(反相矽膠柱的矽膠粒徑為10μm,高徑比為5:1,流速為0.015倍柱體積/min,柱高與較高純度的維生素K2濃縮液上液高度比為7:1)分離,然後用甲醇洗脫並分段收集,即可獲得高純度的維生素K2(純度大於80%);
(6)維生素K2晶體的獲取
以分析純的甲醇將維生素K2於40℃高溫溶解,-40℃低溫結晶,並重結晶,即可獲得維生素K2晶體(純度大於或等於98.2%)。
優選地,所述濃縮方法為減壓濃縮,所述減壓濃縮的溫度為40℃,真空度小於-0.08MPa。
實施例2
一種分離純化納豆芽孢桿菌中維生素K2的方法,如圖1所示,包括如下步驟:
(1)維生素K2萃取液的獲取
以納豆芽孢桿菌發酵液為維生素K2提取原液,離心獲取菌體,烘乾後,採用甲苯進行萃取以獲取維生素K2萃取液;其中萃取方式為靜置萃取,萃取時間60min,萃取次數為3次。
(2)維生素K2上柱原液的獲取
將維生素K2萃取液通過膜分離設備(膜孔徑≤0.45μm)除去不溶性雜質,將獲得的維生素K2濾液減壓蒸乾,然後以丙酮重溶,獲取維生素K2上柱原液;
(3)低純度的維生素K2濃縮液的獲取
將維生素K2上柱原液通過非極性大孔吸附樹脂柱吸附,採用幹法上柱(上柱條件為:高徑比為10:1,流速為0.110倍柱體積/min),然後以極性有機溶劑清洗(用量為大孔吸附樹脂柱柱體積的3倍),最後用異戊烷進行洗脫(用量為大孔吸附樹脂柱柱體積的4倍),收集相應洗脫液並對其進行濃縮,從而獲取低純度的維生素K2濃縮液(純度小於50%);
(4)較高純度的維生素K2濃縮液的獲取
將低純度的維生素K2濃縮液經過分子量排阻層析柱純化,然後以三氯代苯進行洗脫(其用量為分子量排阻層析柱柱體積的4倍),收集相應洗脫液並對其進行濃縮,從而獲取較高純度的維生素K2濃縮液(純度為50-80%);
其中,所述分子量排阻層析填料所排阻的物質的分子量為400Da-1000Da;所述填料可分別做柱或分幾段裝於同一個分子量排阻層析柱中,層析液按照填料排阻極限由小到大或由大到小的順序依次經過柱填料;填充得到的分子量排阻層析柱高徑比為25:1,流速為0.010倍柱體積/min,層析柱高度與維生素K2濃縮液上液高度比為50:1;
(5)高純度的維生素K2的獲取
將較高純度的維生素K2濃縮液溶解於丙酮後,通過反相矽膠柱(反相矽膠柱的矽膠粒徑為80μm,高徑比為25:1,流速為0.065倍柱體積/min,柱高與較高純度的維生素K2濃縮液上液高度比為30:1)分離,然後用丙酮洗脫並分段收集,即可獲得高純度的維生素K2(純度大於80%);
(6)維生素K2晶體的獲取
以含有30%超純水的甲醇將維生素K2於85℃高溫溶解,0℃低溫結晶,並重結晶,即可獲得維生素K2晶體(純度大於98.2%)。
優選地,所述濃縮方法為減壓濃縮,所述減壓濃縮的溫度為99℃,真空度小於-0.08MPa。
實施例3
一種分離純化納豆芽孢桿菌中維生素K2的方法,如圖1所示,包括如下步驟:
(1)維生素K2萃取液的獲取
以納豆芽孢桿菌發酵液(組成為:大豆粉65g/L,玉米粉60g/L,蛋白腖50g/L,發酵周期為:7-9天)為維生素K2提取原液,離心獲取菌體,烘乾後,按照以下步驟提取:
①稱取1.0g幹菌體若干份於50ml的離心管中,分別加入5ml不同的有機溶劑靜置萃取50min,萃取2次,做3個平行樣;其中不同的有機溶劑分別為甲醇、乙醇、1,3丙二醇、正丁醇、乙酸、二氯甲烷、異戊醇、正己烷、乙酸乙酯、三氯甲烷、四氯化碳、丙酮、乙醚、甲苯;
②將獲得的萃取液定容至5ml,獲得檢測液;
③用HPLC法(流動相:甲醇:二氯甲烷=4:1,流速1mL/min,檢測波長248nm)分別檢測各萃取液中維生素K2的濃度含量,其中圖3為以正丁醇為萃取劑時發酵樣品中維生素K2的HPLC液相圖譜圖,圖2為繪製而成的不同有機溶劑萃取維生素K2效率圖,其中甲醇與1,3丙二醇的萃取效率極低,後期試驗中將不作為相應萃取劑的選擇;
(2)維生素K2上柱原液的獲取
將維生素K2萃取液通過膜分離設備(膜孔徑≤0.45μm)除去不溶性雜質,將獲得的維生素K2濾液減壓蒸乾,然後以甲醇重溶,獲取維生素K2上柱原液;
(3)低純度的維生素K2濃縮液的獲取
①稱取一定量的大孔吸附樹脂,以95%乙醇浸泡過夜後,溼法上柱,並以95%的乙醇衝洗至流出液與蒸餾水的混合液(1:5,V/V)無渾濁為止,然後以2倍柱體積的甲醇衝洗柱子,製成的層析柱高徑比為6:1;
②量取一定量的維生素K2甲醇溶液上柱,以0.0283倍柱體積/min的流速過柱,然後以相同的流速用2倍柱體積的甲醇清洗柱子,之後以相同的流速用2倍柱體積的正己烷洗脫維生素K2;
③重複步驟2兩次,作為平行實驗,獲得第二次、第三次維生素K2洗脫液;
測量各維生素K2洗脫液的體積,用HPLC法(流動相:甲醇:二氯甲烷=4:1,流速1mL/min,檢測波長248nm)檢測各洗脫液中維生素K2的含量及油脂性雜質情況,計算維生素K2的回收率,接著,合併相應洗脫液並其進行濃縮,從而獲取低純度(純度小於50%)的維生素K2濃縮液;
其中,獲得的低純度的維生素K2濃縮液不含或含極少量的油脂類極性雜質(質量分數1000Da)與小分子雜質(分子量<400Da)含量之和≤1%,維生素K2回收率≥98%。;
(5)高純度的維生素K2的獲取
①將反相矽膠(粒徑為50μm)經過預處理後,溼法上柱,然後以2倍柱體積的甲醇衝洗柱子,製成的層析柱高徑比為12:1;
②較高純度的維生素K2濃縮液以適量甲醇重溶,取適量上柱,層析柱高度與維生素K2濃縮液上液高度之比為10:1,然後以甲醇進行洗脫,流速為0.0163倍柱體積/min;
③分段收集各截留段即可獲得高純度(純度大於80%)的維生素K2,其中以HPLC法檢測各截留段中維生素K2濃度並計算回收率;
(6)維生素K2晶體的獲取
①對獲得的維生素K2洗脫液進行濃縮,然後以適量的含有10%超純水的甲醇在80℃溫度下重溶;
②將獲得維生素K2甲醇溶液至於-20℃的冰箱中靜置過夜;
③將上述固液混合物4℃低溫過濾,即可獲得維生素K2晶體,並重結晶一次;
④將得到的維生素K2晶體以HPLC上樣液(甲醇:二氯甲烷=4:1)重溶,再採用HPLC法(流動相:甲醇:二氯甲烷=4:1,流速1mL/min,檢測波長248nm)檢測維生素K2晶體的濃度含量,其檢測圖如圖4所示,檢測發現,其純度達到98.2%。
優選地,上述濃縮方法均為減壓濃縮,溫度為40-99℃,真空度小於-0.08MPa。
以上所述僅為本發明的較佳實施例而已,並不用以限制本發明,凡在本發明的精神和原則之內所作的任何修改、等同替換和改進等,均應包含在本發明的保護範圍之內。