一種光催化劑氧化石墨相氮化碳及其製備方法與流程
2023-07-10 18:04:18 2
本發明涉及光催化領域,特別涉及一種製備光催化劑氧化石墨相氮化碳的方法。
背景技術:
光催化技術作為一種「綠色」技術,其在治理水汙染方面,有著與傳統治理水汙染技術不可比擬的很多的優點:(1)操作簡便,耗能較低;(2)光催化反應一般在常溫常壓條件下就可進行,所需的反應條件溫和,而且無機以及有機汙染物能夠被部分或者完全降解,從而使很多的環境汙染物降解生成H2O和CO2,不會產生二次汙染;(3)可以利用太陽光作為光源;(4)有些光催化劑成本低,低毒甚至無毒,穩定性高並且可以重複利用。光催化技術不僅可以用於處理水汙染的問題,而且還可以用於處理大氣汙染、土壤汙染、殺菌等多個方面,光催化技術顯示出了極其廣闊的應用價值。
石墨相氮化碳以其光催化活性較高、穩定性好、原料價格便宜、尤其是不含金屬這一突出優點,使它成為一種新型的光催化劑,但是,單一相催化劑通常因量子效率較低而使其光催化性能表現的不夠理想。
體相石墨相氮化碳材料光生電子-空穴複合率較高,導致其催化效能較低,限制了它在光催化方面的應用。
因此,亟待開發一種光生電子-空穴複合率較低的石墨相氮化碳材料。
技術實現要素:
為了解決上述問題,本發明人進行了銳意研究,結果發現:用碳氮源焙燒製得的石墨相氮化碳,加入氧化劑進行氧化,再除去氧化劑,最終製得的氧化石墨相氮化碳對有機染料的光催化降解效率顯著提高,從而完成了本發明。
本發明的目的在於提供以下方面:
第一方面,本發明提供一種光催化劑氧化石墨相氮化碳,其特徵在於,所述光催化劑根據其XRD光譜,在2θ=12°,27°處存在衍射峰;
根據其紅外光譜,在波數為3300cm-1~3600cm-1,在波數為1200cm-1~1800cm-1和波數為810cm-1處存在特徵吸收帶。
第二方面,本發明還提供一種製備上述光催化劑的方法,其特徵在於,該方法包括以下步驟:
步驟1,將碳氮源進行焙燒,製得石墨相氮化碳;
步驟2,將步驟1製得的石墨相氮化碳加入氧化劑中,反應一定時間後除去氧化劑。
第三方面,本發明還提供上述光催化劑在治理染料汙水,特別是含有有機染料汙水方面的應用,其中,所述有機染料優選為甲基橙、羅丹明B等。
附圖說明
圖1示出樣品的XRD光譜圖;
圖2示出樣品的紅外光譜圖;
圖3示出樣品的光致發光光譜圖;
圖4示出樣品的紫外-可見漫反射光譜圖;
圖5示出樣品在催化降解羅丹明B的可見光催化活性;
圖6示出樣品在催化降解甲基橙的可見光催化活性;
圖7示出清除劑對催化劑催化活性的影響。
具體實施方式
下面通過對本發明進行詳細說明,本發明的特點和優點將隨著這些說明而變得更為清楚、明確。
以下詳述本發明。
根據本發明的第一方面,提供一種光催化劑氧化石墨相氮化碳,其特徵在於,所述光催化劑根據其XRD光譜,在2θ=12°,27°處存在衍射峰;
根據其紅外光譜,在波數為3300cm-1~3600cm-1,在波數為1200cm-1~1800cm-1和波數為810cm-1處存在特徵吸收帶。
根據本發明的第二方面,還提供一種製備上述光催化劑的方法,其特徵在於,該方法包括以下步驟:
步驟1,將碳氮源進行焙燒,製得石墨相氮化碳。
在本發明中,所述碳氮源是一種含氮有機物,即,同時含有氮元素及碳元素的小分子有機物,特別是指在加熱條件下能夠分解的含氮小分子有機物,其在製備石墨相氮化碳中既作為氮源物質又作為碳源物質。
在本發明中,所述碳氮源為碳氮比為1:3~3:1的小分子量的含氮有機物,優選碳氮比為1:2的小分子量含氮有機物,如單氰胺、二氰二胺、三聚氰胺、尿素、鹽酸胍等,優選為二氰二胺。
在本發明中,將碳氮源在400℃~800℃下焙燒時能夠生成石墨相氮化碳,而且製得的產物形貌均一,本發明優選焙燒溫度為400℃~800℃,更優選為450℃~600℃,如550℃。
本發明人進一步發現,焙燒時間為1~5小時即可充分反應,因此,本發明優選選擇焙燒時間為1~5小時,優選為1.5~4小時,更優選為2~3.5小時,如2小時。
在本發明中,任選地,將焙燒後的物質進行冷卻,並進行粉碎。
步驟2,將步驟1製得的石墨相氮化碳加入氧化劑中,反應一定時間後除去氧化劑。
在本發明中,所述氧化劑為濃硫酸與重鉻酸鉀的組合物。
在本發明中,所述濃硫酸為重量百分數為95%以上的濃硫酸,優選為重量百分數為98%以上的濃硫酸。
在本發明中,所述氧化劑中,濃硫酸與重鉻酸鉀的體積重量比為100mL:(10.0~30.0)g,優選為100mL:(15.0~25.0)g,如100mL:20.0g,其中,濃硫酸的體積以濃硫酸溶液的總體積計。
本發明人發現,當濃硫酸與重鉻酸鉀按照上述體積重量比進行組合時,得到的氧化劑具有強氧化性,能夠將化學性質穩定的石墨相氮化碳表面進行氧化,改變石墨相氮化表面的形,不受任何理論的束縛,本發明人認為經過上述氧化劑的改性後,石墨相氮在其表面形成微孔結構,具有吸附性。
在本發明一種優選的實施方式中,所述氧化劑是將重鉻酸鉀固體加入濃硫酸中而製得的。
本發明人發現,將重鉻酸鉀固體加入濃硫酸後,體系溫度升高,顏色加深,優選地,當體系的溫度降低至室溫後再使用。
在本發明中,步驟1製得的石墨相氮化碳在氧化劑中的處理的時間為1~5小時,優選為1.5~3小時,如2小時。
本發明人發現,將石墨相氮化碳加入所述氧化劑後,體系再次放出大量的熱,利用體系釋放出的熱量增加石墨相表面被氧化速度和程度,隨著體系溫度的降低,石墨相氮化碳表面的氧化過程完成。
在本發明中,當體系溫度降低至室溫後,將體系倒入蒸餾水中,當體系的溫度降低至室溫後除去體系中的液相。
在本發明中,除去體系中液相的方式為常壓過濾法、減壓過濾法或者為離心法,優選為離心法。
除去液相後,將得到的固體進行洗滌,優選使用蒸餾水進行洗滌,以除去無機雜質。
任選地,將洗滌後的固體進行乾燥。
在本發明一種優選的實施方式中,乾燥的溫度為60℃~120℃,優選為70℃~110℃,如80℃。
優選地,乾燥的時間為8~20小時,優選為10~18小時,如15小時。
根據本發明的第三方面,還提供上述光催化劑在治理染料汙水,特別是含有有機染料汙水方面的應用,其中,所述有機染料優選為甲基橙、羅丹明B等。
根據本發明提供的光催化劑氧化-石墨相氮化碳及其製備方法,具有以下有益效果:
(1)本發明提供的方法操作簡便易行,易於實現,製備光催化劑的生產效率高;
(2)本發明提供的方法綠色環保,無汙染產生;
(3)本發明提供的光催化劑光催化效率高;
(4)本發明提供的光催化劑光催化性能穩定,可重複使用。
實施例
實施例1
(1)準確稱取二氰二胺6.0g,置於清洗乾淨的坩堝中,蓋上蓋子,然後將其於550℃馬弗爐中焙燒4h,焙燒之後於瑪瑙研體中研磨,然後裝入袋子中密封好,即可製得g-C3N4光催化劑固體粉末,標記為Bulkg-C3N4。
(2)稱取步驟1製得的光催化劑粉末1.000g,用100mL量筒量取濃度為98%的H2SO4100mL於250mL的燒杯中,用天平稱取K2Cr2O7固體20.000g加入上述H2SO4溶液中,然後立刻使用磁力攪拌器攪拌,在混合溶液顏色變為棕色後,將上述稱量好的g-C3N4光催化劑粉末放入混合溶液中,攪拌2h至體系降至室溫。
將冷卻的混合物慢慢的倒入800mL蒸餾水中,冷卻至室溫,再將所得液體在高速冷凍離心機中離心(6000rpm),得到化學氧化後的g-C3N4固體,將所得固體多次抽濾、清洗去除固體表面殘留的酸,然後80℃恆溫蒸發乾燥得淡黃色固體,淡黃色固體即為化學氧化g-C3N4光催化劑,於透明小袋中密封保存(標記為化學氧化g-C3N4)。
對比例
對比例1
本對比例所用樣品為實施例1步驟1中製得的C3N4。
實驗例
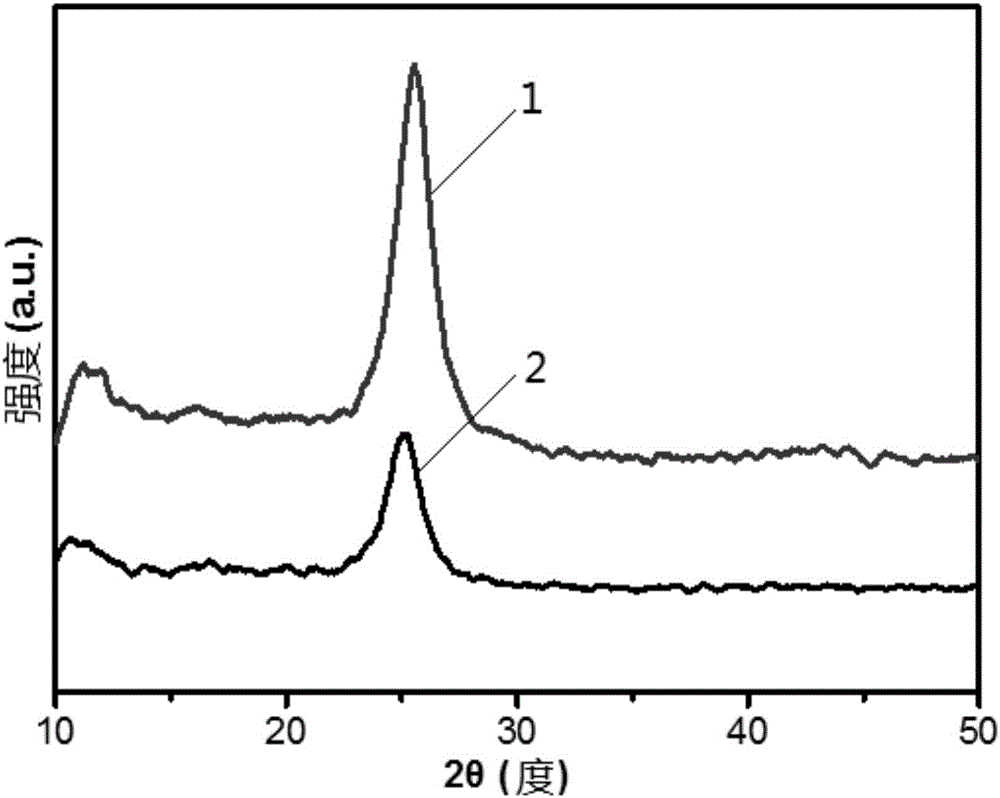
實驗例1樣品的XRD光譜分析
本實驗例所用樣品為實施例1和對比例1製得。
採用Bruker D8 Advance型X射線衍射儀(XRD),銅靶(Cu Kα(λ=0.154nm))射線,Ni濾光片,工作電壓40kV,電流40mA,掃描範圍2θ=10°~60°,分析樣品的晶相結構,結果如圖1所示,其中,
曲線1示出實施例1製得樣品的XRD光譜圖;
曲線2示出對比例1製得樣品的XRD光譜圖。
在衍射角2θ=12°,27°處的衍射峰分別對應於g-C3N4的(100)和(002)晶面,是辨認石墨相g-C3N4的特徵衍射峰。
由圖1可知,對比例1製得樣品的XRD譜圖中存在上述衍射峰,說明對比例1製備的光催化劑是石墨相g-C3N4。
由圖1還可知,實施例1製得的光催化劑X射線衍射峰值強於對比例1製得樣品的X射線衍射峰值,這表明實施例1製得樣品的結晶度相比於對比例1製得樣品要好,這還表明化學氧化改變了石墨相g-C3N4的結構,增大了其自身的結晶度,同時大大增強其光催化活性,這與後續實驗測定的光催化活性是一致的。
實驗例2樣品的紅外光譜分析
本實驗例所用樣品為實施例1和對比例1製得。
取少量上述粉末樣品,分別加入少量溴化鉀粉末,研磨使樣品與溴化鉀混合均勻,壓成薄片,用傅立葉變換紅外光譜儀對催化劑進行紅外光譜表徵,結果如圖2所示,其中,
曲線1示出實施例1製得樣品的紅外光譜圖;
曲線2示出對比例1製得樣品的紅外光譜圖。
由圖2可知,實施例1和對比例1製得的光催化劑的紅外光譜圖分別在波數為3300cm-1~3600cm-1,在波數為1200cm-1~1800cm-1和波數為810cm-1處出現明顯的特徵吸收帶,其中,在波數為3300cm-1~3600cm-1範圍內的寬波段為N-H鍵的伸縮振動吸收帶,在波數為1200cm-1~1800cm-1範圍內的幾個強的波段是CN雜環化合物的結構特徵峰在810cm-1附近出現吸收帶,歸屬於3-S三嗪的特徵峰,這標誌著g-C3N4的生成。
由圖2可知,對比例1製得樣品和實施例1製得樣品的紅外吸收譜圖基本相似,這表明兩種樣品在化學結構上相同或者基本相似。
實驗例3樣品的光致發光光譜分析
本實驗例所用樣品為實施例1和對比例1製得。
取少量上述光催化劑粉末樣品,利用螢光光譜儀測試各種催化劑樣品的光致發光性能。實驗中,應儘可能用玻片將樣品壓得緻密,以保持樣品表面的平整,且一個樣品應至少平行測試兩次,保證數據的有效性,結果如圖3所示,其中,
曲線1示出實施例1製得樣品的光致發光光譜圖;
曲線2示出對比例1製得樣品的光致發光光譜圖。
光致發光光譜能夠揭示光生載流子的遷移、捕獲和複合等規律,常用來研究光催化劑中電子和空穴的捕獲、累積或轉移,當光生電子和空穴複合時,部分能量以光的形式釋放出來,發出螢光,弱螢光強度意味著電子空穴複合概率有所降低。
圖3為樣品用300nm光激發後的螢光發射光譜,由圖3可知,兩種樣品均出現很強的螢光發射峰,這與對比例1製得樣品的禁帶寬度(2.78eV)相一致,即電子受激發後發生從n到π的躍遷,之後回到基態與空穴結合,產生螢光。
由圖3還可知,實施例1製得樣品的螢光強度低,說明其光生電子-空穴的複合機率低,其光催化活性高。
實驗例4樣品的紫外-可見漫反射光譜分析
本實驗例所用樣品為實施例1和對比例1製得。
首先打開儀器,進行自檢,然後進行基線校正(兩個都放水參比),從測量中,進行參數設置,設置好後,進行基線校正。待基線校正好後,取少量上述光催化劑粉末樣品,對各樣品分別研磨後,利用雙光束紫外-可見分光光度計對各種催化劑樣品進行表徵。實驗中,對樣品研磨細緻,且在壓片時應儘可能將樣品壓得緻密,以保持樣品表面的平整,測試波長範圍200-800nm,結果如圖4所示,其中,
曲線1示出實施例1製得樣品的紫外-可見漫反射光譜圖;
曲線2示出對比例1製得樣品的紫外-可見漫反射光譜圖。
圖4顯示出對比例1製得樣品的最大吸收波長約為400nm,實施例1製得樣品的最大吸收波長約為380nm。
由圖4可知,實施例1製得樣品和對比例1製得樣品相比,吸收帶邊沿有明顯的藍移,另外,與對比例1製得光催化劑相比,實施例1製得樣品有著較高的紫外光吸收峰,這可能是由於光催化劑的比表面積增大,改變了層間的電子耦合,從而增強了光催化活性,這也與實驗測定的光催化活性結果是一致的。
實驗例5樣品的可見光催化活性分析
本實驗例所用樣品為實施例1和對比例1製得。
分別準確稱取0.025g上述光催化劑粉末於石英管中,編號1、2。然後向兩個石英管中各加入50mL濃度為10mg/L的羅丹明B溶液,再各加入一個磁力攪拌子,將石英管放入光化學反應儀中,先開啟循環水冷卻裝置再打開電源,在連續攪拌下,暗反應處理100min後,取樣離心(14000rpm)20min分別測其吸光度記為A1和B1,然後開光源300W氙燈光照,每隔20分鐘取樣離心分別測其吸光度記為AX和BX,然後分別計算降解率WA(%)=(A1-AX)/A1×100%和WB(%)=(B1-BX)/B1×100%。然後根據所得數據繪製(降解率—時間)可見光降解活性圖,如圖5所示,其中,
曲線1示出實施例1製得樣品催化降解羅丹明B的可見光活性結果;
曲線2示出對比例1製得樣品催化降解羅丹明B的可見光活性結果。
由圖5可知,在可見光照射60min後,對比例1製得催化劑光催化降解羅丹明B降解率達到51.3%,然而實施例1製得催化劑光催化降解羅丹明B降解率達到92.5%,兩者相比較,實施例1製得光催化劑的催化性能提高了41.2%。
另分別準確稱取0.025g的對比例1和實施例1製得的光催化劑粉末於石英管中,編號1、2。然後向兩個石英管中各加入50mL濃度為10mg/L的甲基橙溶液,其餘步驟同羅丹明B實驗,做平行實驗,然後根據所得數據繪製(降解率—時間)可見光降解活性圖,結果如圖6所示,其中,
曲線1示出實施例1製得樣品催化降解甲基橙的可見光活性結果;
曲線2示出對比例1製得樣品催化降解甲基橙的可見光活性結果。
由圖6可知,在可見光照射60min後,對比例1製得催化劑光催化降解甲基橙的降解率達到32.4%,然而實施例1製得催化劑光催化降解甲基橙的降解率達到98.5%,兩者相比較,實施例1製得催化性能提高了66.1%。
由上述的兩個實驗數據對比可以看出與對比例1製得樣品相比,實施例1製得光催化劑光催化活性有著明顯的提高。
實驗例6清除劑對樣品催化活性的影響
本實驗以羅丹明B為模型化合物,通過引入各種自由基清除劑,研究光催化劑的光催化機制。
本實驗例所用樣品為實施例1製得。
準確稱取5份0.025g實施例1製得樣品的粉末於5個石英管中,編號1、2、3、4、5,依次向石英管中加入50mL濃度為10mg·L-1的羅丹明B溶液。
在1號管中不加入任何清除劑(記為No scavenger),
在2號管中加入5.000μL異丙醇(IPA),起到抑制體系降解過程中·OH產生的作用
在3號管中加入0.004g草酸銨(AO),起到抑制體系降解過程中h+產生的作用
在4號管中加入0.004g對苯醌(BQ),起到抑制體系降解過程中·O2-產生的作用
在5號管中加入3.800μL過氧化氫酶(CAT)起到抑制體系降解過程中H2O2產生的作用。
每個石英管中均加入一個磁力攪拌子。將石英管放入光化學反應儀中,在持續攪拌下,暗處理100min,取樣離心,分別測其吸光度A0。打開光源,紫外光照處理1h,取樣離心,測其吸光度AX,計算降解率W(%)=(A0-AX)/A0×100%,可得不同清除劑對催化劑可見光活性的影響程度。並繪製成圖。
由圖7可以看出,在其他的條件不變的情況下,(1)與不添加清除劑相比較,加入清除劑後,實施例製得的光催化劑的活性均有所降低;(2)在其他條件相同的情況下,對苯醌(BQ)的加入對催化劑見光光催化活性的影響最大,說明在可見光照射下光催化降解羅丹明B的過程中·O2-是最主要的活性物種;也就是說,·O2-、·OH、H2O2和h+在光催化降解過程中起明顯作用,尤其是·O2-在光催化過程中起最主要作用。
以上結合具體實施方式和範例性實例對本發明進行了詳細說明,不過這些說明並不能理解為對本發明的限制。本領域技術人員理解,在不偏離本發明精神和範圍的情況下,可以對本發明技術方案及其實施方式進行多種等價替換、修飾或改進,這些均落入本發明的範圍內。本發明的保護範圍以所附權利要求為準。