一種RP‑HPLC測定兩面針中藥牙膏中毛兩面針素含量的方法與流程
2023-06-03 02:56:59 4
本發明屬於化學檢測領域,尤其是一種rp-hplc測定兩面針中藥牙膏中毛兩面針素含量的方法。
背景技術:
兩面針,為芸香科植物兩面針zanthoxylumnitidum(roxb.)dc.的乾燥根,可活血化瘀,行氣止痛,祛風通絡,解毒消腫,不能過量服用[1],毛兩面針為其變種[2],兩面針藥材在化學成分及含量上均有別於毛兩面針。毛兩面針素又名飛龍掌血內酯,為毛兩面針及飛龍掌血根特有成分;氯化兩面針鹼為兩面針主要效用成分之一;存在於藥材兩面針、毛兩面針及飛龍掌血中,但在三者中含量有所不同[3-5]。《中國藥典》2015版一部兩面針的檢查項下明確規定不得檢出毛兩面針素。
兩面針中藥牙膏是添加中藥兩面針提取物作為日常維護口腔健康的中藥牙膏,具有消炎止血,鎮痛抗菌,清熱降火的功效[6]。用於牙齦出血,牙齦腫痛。
毛兩面針素在兩面針牙膏中的含量緊緊關聯著兩面針牙膏的質量和有效性,有研究發現藥材市場上主流藥材並非藥典所規定品種,而以毛兩面針為主,可能出現用毛兩面針藥材替代兩面針投料的情況[3]。儘管目前有研究發現對毛兩面針素含量極高的飛龍掌血藥材和毛兩面針素對照品進行急性毒性評價,未觀察到毒性反應[7],但目前並沒有研究表明毛兩面針或飛龍掌血可替代兩面針投料,為了保證添加功效成分來源的純正性,兩面針中藥牙膏仍應不得檢出毛兩面針素。目前對兩面針及毛兩面針藥材主要成分的含量測定[3]與牙膏中有效成分的含量測定[4]已有相關研究報導,但尚未見有對兩面針牙膏中毛兩面針素的含量測定方法進行報導。
由藥典[1]可知,兩面針的檢查項下明確規定不得檢出毛兩面針素,而在本實驗三批樣品中毛兩面針素含量的結果(見表6)發現其中一批樣品中檢出微量的毛兩面針素,為不合格樣品。其餘兩批不同樣品皆為添加了兩面針提取物的中藥牙膏,在含量測定項下的結果顯示未檢出毛兩面針素,原因可能為不含毛兩面針素或毛兩面針素含量極微並低於檢出限。
目前,兩面針的質量控制指標尚未完善,在製備兩面針中藥牙膏時選用的兩面針藥材的質量會有所差別,但兩面針牙膏配方工藝嚴格保密,其質量標準也未見有相關的報導,所以在原料藥兩面針的選取上也會對兩面針牙膏中兩面針功效成分的含量及真偽造成影響。
因此建立反相高效液相色譜法測定兩面針中藥牙膏中毛兩面針素的含量對控制該牙膏的質量和保證其日常使用的有效性、安全性有著重要的意義。
技術實現要素:
針對上述不足,本發明旨在提供一種rp-hplc測定兩面針中藥牙膏中毛兩面針素含量的方法,該方法具有操作簡便、迅速、準確、幹擾少、精密度與重複性較好等優點,可為測定兩面針中藥牙膏中毛兩面針素的含量提供實驗依據。
為了解決上述技術問題,本發明提供的技術方案是這樣的:一種rp-hplc測定兩面針中藥牙膏中毛兩面針素含量的方法,依次包括下述步驟:
步驟1:採用甲醇提取兩面針牙膏中的毛兩面針,並收集甲醇提取液;
步驟2:採用rp-hplc法測定甲醇提取液中毛兩面針的含量。
優選地,所述的步驟1具體包括下述步驟:
步驟s1:稱取兩面針牙膏2g,與20ml濃度為80%的甲醇混合,稱定重量m1;
步驟s2:攪拌30min,稱定重量,並用濃度為80%的甲醇補足至m1;
步驟s3:超聲30min,冷卻,稱定重量,並用濃度為80%的甲醇補足至m1,離心,冷藏靜置10min;
步驟s4:取上清液1.5-2ml,離心5min,過濾,即得。
優選地,步驟s3所述的超聲處理的參數為100w、80hz。
優選地,步驟s3所述的離心參數為:4000r/min,5min。
優選地,步驟s4所述的離心參數為:15000r/min,5min。
優選地,步驟s4所述的過濾具體為:用0.22μm微孔濾膜過濾。
優選地,步驟2所述的rp-hplc的檢測參數為:色譜柱為agilentzorbaxsb-c18;流動相為0.1%甲酸水溶液-乙腈;流速為1.0ml/min;進樣量為1μl;柱溫為35℃;檢測波長為328nm。
優選地,所述的色譜柱的規格為4.6mm×250mm,5μm。
優選地,所述的流動相為體積比等於6:4的0.1%甲酸水溶液-乙腈。
優選地,所述的流動相採用等梯度洗脫,洗脫時間為6min。
本發明與傳統方法相比,具有以下優點:
(1)兩面針中藥牙膏中的兩面針提取物主要成分為氯化兩面針鹼及可能由兩面針主要偽品毛兩面針及飛龍掌血替代投料產生的毛兩面針素,根據毛兩面針素以及氯化兩面針鹼分子結構(圖3)中有一定的極性基團[8-9],所以比較了極性較低的zorbaxsb-c18(4.6mm×250mm,5μm)及welchutimate@hplcaq-c18(2.1mm×50mm,1.8μm),檢測結果顯示,兩種低極性色譜柱中,使用長柱zorbaxsb-c18(4.6mm×250mm,5μm)對毛兩面針素分離效果較好,幹擾成分少,峰面積較高,該色譜柱鍵合耐溶劑衝洗的固定相,對複雜的高極性的化合物具有優異的分離能力,因此本發明選用zorbaxsb-c18(4.6mm×250mm,5μm)色譜柱。
(2)本發明考察了多種流動相體系,包括0.02mol/l乙酸銨-乙腈、0.05mol/l磷酸二氫鈉-磷酸-乙腈、0.1%甲酸水-乙腈,結果表明:採用0.1%甲酸水-乙腈系統作為流動相,分離效果較好,不出現拖尾現象,且流動相為弱酸性,對色譜柱損害較小,毛兩面針素及氯化兩面針鹼對照品均在273nm、328nm處發現最大吸收波長,後者峰形較良好。故採用0.1%甲酸水-乙腈作為流動相,使用dad檢測器,選取328nm作為檢測波長,等梯度梯度洗脫測定毛兩面針素含量。
(3)由於毛兩面針素易溶於極性較大的有機溶劑甲醇[9],牙膏中的其他主要組成部分如粘合劑(纖維素衍生物及其他物質)與摩擦劑等不溶於有機溶劑,但如果在膏體中直接加入甲醇,則膏體會凝聚成團,被測成分被包裹在體內而難以溶出,故本文分別不同體積分數的70%甲醇、75%甲醇、80%甲醇、85%甲醇、90%甲醇溶液對樣品進行提取,其他處理方法不變,結果分析表明,用80%甲醇作為提取溶劑,毛兩面針素峰面積明顯比其餘不同體積分數的溶劑所得峰面積要大,且具有良好峰型,所以選用80%甲醇作為提取溶劑。
(4)參考文獻中牙膏有效成分的提取一般為磁力攪拌及超聲各30min,水浴回流30min等,以毛兩面針素的含量為指標,分別考察比較兩種提取方法,方法分別為:①磁力攪拌30min+超聲30min、②磁力攪拌30min+水浴回流30min,其他處理條件不變。結果表明:兩者待測成分提取量相差不大,但水浴回流效果較差。分析原因可能在於牙膏不易分散均勻,待測物溶出不足,水浴回流需要多次轉移樣品溶液易造成損失,操作不夠簡便,故排除水浴回流選用方法①作為提取方法。
(5)在選用提取方法為磁力攪拌30min條件下,進一步比較了超聲20min、超聲40min、超聲50min、超聲60min與超聲30min三者的峰面積的差別,結果顯示,超聲20min得到的峰面積較小,超聲40min、超聲50min、超聲60min所得的峰面積皆較超聲30min小,故確定選用超聲30min作為提取方法。
(6)本發明所述的測定方法具有操作簡便、迅速、準確、幹擾少、精密度與重複性較好等優點,可為測定兩面針中藥牙膏中毛兩面針素的含量提供實驗依據。
附圖說明
圖1為毛兩面針素及氯化兩面針鹼混合標準對照溶液的色譜圖;
圖2為毛兩面針素對照品溶液的色譜圖;
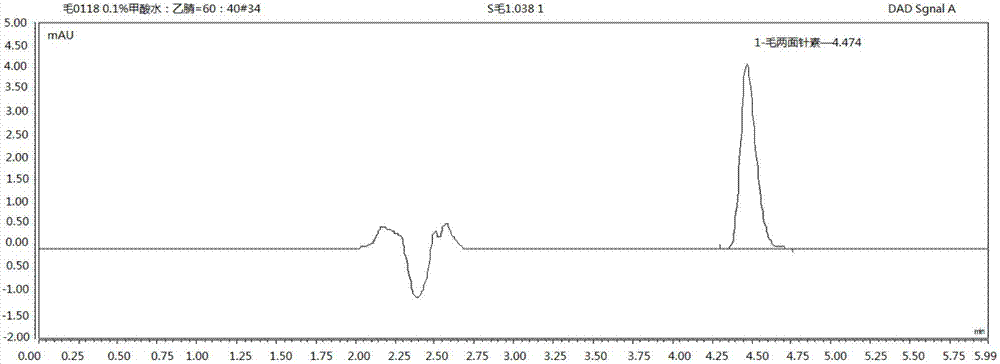
圖3為供試品溶液的色譜圖;
圖4為加標供試品溶液的色譜圖;
圖5為陰性對照的色譜圖;
圖6為毛兩面針素濃度(μg/ml)-峰面積線性回歸曲線圖;
圖7為毛兩面針素及氯化兩面針鹼分子結構式;
圖8為提取溶劑選擇的比較結果圖。
具體實施方式
下面結合具體實施方式對本發明的權利要求書做進一步詳細說明,但不構成對本發明的任何限制,任何在本發明的權利要求保護範圍內所做的任何修改、等同替換仍屬於本發明所要保護的範圍內。
實施例1
1儀器與試藥
1.1儀器
安捷倫生物惰性四元液相色譜儀(儀器型號:agilent1260;廠家:agilenttechnology);
二極體陣列檢測器(dad);
電子分析天平(儀器型號:cpa225d;廠家:賽多利斯科學儀器(北京)有限公司);
超聲波清洗器(儀器型號:elmasonicp180h;廠家:德國elmaschmidbauergmbh);
超純水儀(儀器型號:elgapurelabclassicuvf;廠家:法國威立雅集團);
加熱磁力攪拌器(儀器型號:c-maghs7;廠家:德國ika);
高性能通用臺式離心機(儀器型號:allegrax-15r;廠家:美國貝克曼公司);
臺式冷凍離心機(儀器型號:3k15;廠家:德國sigma)。
1.2試藥
毛兩面針素對照品(由中國食品藥品檢定研究院提供,批號為200001,供含量測定用);
氯化兩面針鹼對照品(中國食品藥品檢定研究院批號110848-200502,供含量測定用);
兩面針中藥牙膏樣品(由某製藥公司提供,產品批號分別為b20190216a1、g20190524b1、f20190612b1,本文中方法學採用產品批號為b20190216a1的兩面針牙膏樣品);
甲醇(omnichem色譜純);
乙腈(omnichem色譜純);
甲酸(廣東光華科技股份有限公司分析純);
超純水。
2實驗方法及結果
2.1色譜條件
色譜柱:採用agilentzorbaxsb-c18(4.6mm×250mm,5μm);
流動相a為0.1%甲酸水溶液,流動相b為乙腈,等梯度洗脫6min,a為60%,b為40%;
流速:1.0ml/min;
進樣量1μl;
柱溫35℃;
檢測波長為328nm;
此色譜條件下氯化兩面針鹼與毛兩面針素可很好分離,幹擾性小;理論塔板數按氯化兩面針鹼峰計算,應不低於5000,待測組分與相鄰共存物之間的分離度應大於1.5。
2.2對照品與供試品的製備
2.2.1對照品溶液的製備
精密稱取毛兩面針素對照品適量,加入80%甲醇配製成每1ml含0.1036μg的溶液;另精密稱取氯化兩面針鹼對照品適量,加入80%甲醇與毛兩面針素對照品配製成每1ml含0.5μg的混合標準對照品溶液。
2.2.2供試品溶液的製備
取供試品約2g,精密稱定,置具塞錐形杯中,精密加入80%甲醇20ml,稱定重量,磁力攪拌30min,稱定重量,超聲處理30min(100w,80hz,下文出現的皆同此),放冷,再稱定重量,後兩次稱定後皆用80%甲醇補足減失的重量,搖勻,將樣品轉移至50ml離心管中,4000r/min離心分離5min,冰櫃冷藏靜置10min,取上清液1.5ml至2ml離心管中,15000r/min離心分離5min,上清液用0.22μm微孔膜濾過即得供試品溶液。
2.3系統適用性考察
按2.1項下的色譜條件對供試品溶液與對照品溶液進行測定,得到相應的色譜圖(見圖1)。結果表明:兩面針牙膏供試品溶液的色譜圖在與毛兩面針素對照品相應保留時間處未出現明顯毛兩面針素的色譜峰,但是對供試品進行加樣(加入毛兩面針素標準對照品溶液15μl,濃度:0.4144mg/ml)同法處理供試品,加樣供試品溶液的色譜圖在與毛兩面針素對照品相應保留時間處出現明顯毛兩面針素的色譜峰,說明該色譜條件適用於毛兩面針素成分的分析與測定。
2.4線性關係考察
取毛兩面針素對照品10.36mg(含量按100%計),精密稱定,加80%甲醇25ml(濃度:0.4144mg/ml)作為儲備液,再精密吸取0.25ml置10ml容量瓶中,加80%甲醇稀釋至刻度(濃度:1.036μg/ml),然後分別精密吸取該溶液0.5ml、1ml、2ml、4ml、8ml置10ml容量瓶中,加入80%甲醇稀釋至刻度,搖勻,並依照上述方法測定,結果詳見表1。
以濃度(μg/ml)-峰面積進行線性回歸,求得回歸方程:y=0.455x+5e-05,r2=0.9997。毛兩面針素在0.0518~0.8288μg/ml範圍內呈良好的線性關係,詳見圖2。
表1線性實驗結果
2.5精密度試驗
取濃度為0.4144μg/ml的對照品溶液,按上述色譜條件連續測定6次,所得平均峰面積為0.0842,rsd=0.35%,詳見表2。
結果表明,本方法的精密度較好。
表2精密度實驗結果
2.6穩定性試驗
取樣品(批號:b20190216a1)約2g,精密稱定,按2.2.2項下的供試品製備方法製備供試品溶液。以1μl為進樣量,測定樣品中毛兩面針素的峰面積。並每隔一定的時間測定一次,共測5次,詳見表3。
結果表明,在10h內,被測物穩定。
表3穩定性實驗結果
2.7重複性試驗
取相同批號的樣品(批號:b20190216a1)2g,共6份,精密稱定,按2.2.2項下的提取方法製備供試品溶液,進樣量為1μl,以上述的色譜條件平行實驗,測得樣品中毛兩面針素的含量為0.1258μg/g,rsd為0.98%。
結果表明本方法重複性良好,結果詳見表4。
表4重複性試驗結果
2.8加樣回收率實驗
精密稱取已知含量兩面針牙膏(批號:b20190216a1,毛兩面針素含量為0.1258μg/g2g,共取6份,置具塞錐形瓶中,精密加入毛兩面針素對照品溶液(濃度為0.4144mg/ml)1.5μl,再精密加入80%甲醇20ml,稱定重量,磁力攪拌30min,稱定重量,超聲處理30min,放冷,再稱定重量,後兩次稱定後皆用80%甲醇補足減失的重量,搖勻,將樣品轉移至50ml離心管中,4000r/min離心分離5min,冰櫃冷藏靜置10min,取上清液1.5ml至2ml離心管中,15000r/min離心分離5min,上清液用0.22μm微孔膜濾過即得供試品溶液。
按上述方法測定,結果顯示本方法回收率較好,詳見表5。
表5回收率實驗結果
2.9含量測定
取本品3批(批號分別為:b20190216a1、g20190524b1、f20190612b1)各2g,精密稱定,按2.2.2項下的方法製備供試品溶液,進樣1μl,測定毛兩面針素的含量,結果詳見表6。
表6含量測定結果(n=3)
3討論
3.1色譜柱及流動相的選擇
兩面針中藥牙膏中的兩面針提取物主要成分為氯化兩面針鹼及可能由兩面針主要偽品毛兩面針及飛龍掌血替代投料產生的毛兩面針素,根據毛兩面針素以及氯化兩面針鹼分子結構(圖3)中有一定的極性基團[8-9],所以比較了極性較低的zorbaxsb-c18(4.6mm×250mm,5μm)及welchutimate@hplcaq-c18(2.1mm×50mm,1.8μm),檢測結果顯示,兩種低極性色譜柱中,使用長柱zorbaxsb-c18(4.6mm×250mm,5μm)對毛兩面針素分離效果較好,幹擾成分少,峰面積較高,該色譜柱鍵合耐溶劑衝洗的固定相,對複雜的高極性的化合物具有優異的分離能力,因此本文中選用zorbaxsb-c18(4.6mm×250mm,5μm)色譜柱。
本實驗考察了多種流動相體系,包括0.02mol/l乙酸銨-乙腈、0.05mol/l磷酸二氫鈉-磷酸-乙腈、0.1%甲酸水-乙腈,結果表明:採用0.1%甲酸水-乙腈系統作為流動相,分離效果較好,不出現拖尾現象,且流動相為弱酸性,對色譜柱損害較小,毛兩面針素及氯化兩面針鹼對照品均在273nm、328nm處發現最大吸收波長,後者峰形較良好。故採用0.1%甲酸水-乙腈作為流動相,使用dad檢測器,選取328nm作為檢測波長,等梯度梯度洗脫測定毛兩面針素含量。
3.2提取溶劑的選擇
由於毛兩面針素易溶於極性較大的有機溶劑甲醇[9],牙膏中的其他主要組成部分如粘合劑(纖維素衍生物及其他物質)與摩擦劑等不溶於有機溶劑,但如果在膏體中直接加入甲醇,則膏體會凝聚成團,被測成分被包裹在體內而難以溶出,故本文分別不同體積分數的70%甲醇、75%甲醇、80%甲醇、85%甲醇、90%甲醇溶液對樣品進行提取,其他處理方法不變,結果分析表明,用80%甲醇作為提取溶劑,毛兩面針素峰面積明顯比其餘不同體積分數的溶劑所得峰面積要大,且具有良好峰型,所以選用80%甲醇作為提取溶劑,比較結果詳見圖4。
3.3提取方法的選擇
參考文獻中牙膏有效成分的提取一般為磁力攪拌及超聲各30min,水浴回流30min等,以毛兩面針素的含量為指標,分別考察比較兩種提取方法,方法分別為:①磁力攪拌30min+超聲30min、②磁力攪拌30min+水浴回流30min,其他處理條件不變,結果比較詳見表8,兩者待測成分提取量相差不大,但水浴回流效果較差。分析原因可能在於牙膏不易分散均勻,待測物溶出不足,水浴回流需要多次轉移樣品溶液易造成損失,操作不夠簡便,故排除水浴回流選用方法①作為提取方法。
在選用提取方法為磁力攪拌30min條件下,進一步比較了超聲20min、超聲40min、超聲50min、超聲60min與超聲30min三者的峰面積的差別,結果顯示,超聲20min得到的峰面積較小,超聲40min、超聲50min、超聲60min所得的峰面積皆較超聲30min小,故確定選用超聲30min作為提取方法,詳見表9。
表8不同提取方法結果比較
表9不同超聲時間的結果比較
3.4含量測定結果分析
目前,兩面針的質量控制指標尚未完善,在製備兩面針中藥牙膏時選用的兩面針藥材的質量會有所差別,但兩面針牙膏配方工藝嚴格保密,其質量標準也未見有相關的報導,所以在原料藥兩面針的選取上也會對兩面針牙膏中兩面針功效成分的含量及真偽造成影響。
由藥典[1]可知,兩面針的檢查項下明確規定不得檢出毛兩面針素,而在本實驗三批樣品中毛兩面針素含量的結果(見表6)發現其中一批樣品中檢出微量的毛兩面針素,為不合格樣品。其餘兩批不同樣品皆為添加了兩面針提取物的中藥牙膏,在含量測定項下的結果顯示未檢出毛兩面針素,原因可能為不含毛兩面針素或毛兩面針素含量極微並低於檢出限。
本文採用高效液相色譜法對兩面針中藥牙膏中毛兩面針素的含量進行測定,該方法具有操作簡便、迅速、準確、幹擾少、精密度與重複性較好等優點,可為測定兩面針中藥牙膏中毛兩面針素的含量提供實驗依據。
以上所述僅為本發明的較佳實施例,凡在本發明的精神和原則範圍內所做的任何修改、等同替換和改進等,均應包含在本發明的保護範圍內。